SurfaceProcessSimulation¶
- class SurfaceProcessSimulation(substrate, filename, object_id, random_seed, temperature, thermostat_method=None, fixed_thickness=<class 'NL.ComputerScienceUtilities.NLFlag._NLFlag.Automatic'>, thermostat_thickness=<class 'NL.ComputerScienceUtilities.NLFlag._NLFlag.Automatic'>, adaptive_thermostat_layer=None, distance_above_surface=None, log_interval=None, trajectory_interval=None, log_filename_prefix=None, active_learning=None)¶
Set up a surface process simulation to study deposition, etching, or sputtering.
- Parameters:
substrate (
BulkConfiguration) – The initial surface (bulk slab model).filename (str) – The full or relative path to save the restart information to.
object_id (str) – The object id to use when saving the restart information.
random_seed (int) – The seed for the random number generator.
temperature (PhysicalQuantity of type temperature) – The thermostat temperature.
thermostat_method (NVTBerendsen | NVTNoseHoover | Langevin) – The class that should be used for the thermostat method in the molecular dynamics simulation. Default: NVTNoseHoover
fixed_thickness (PhysicalQuantity of type length) – The length of the bottom fixed layer of the slab. Default: 10 * Angstrom.
thermostat_thickness (PhysicalQuantity of type length) – The length of the thermostat layer of the slab (above the fixed layer). Default: 10 * Angstrom.
adaptive_thermostat_layer (bool) – Whether to grow (or shrink) the length of the thermostat layer such that the width of the reactive layer remains approximately the same as new layers are deposited (or removed). The length of the reactive layer is taken from the initial configuration (i.e. total_initial_thickness - fixed_thickness - thermostat_thickness) Default: False
distance_above_surface (PhysicalQuantity of type length) – The distance above the surface where new atoms are inserted into the system. Default: 10 * Angstrom.
log_interval (int | PhysicalQuantity of type time) – The frequency with which log messages are written. The value can be given either as a number of MD steps (integer) or as a time interval. Default: 100.
trajectory_interval (int) – The frequency with which configurations are saved in the trajectory. The value can be given either as a number of MD steps (integer) or as a time interval. Default: 100.
log_filename_prefix (str) – An optional prefix to the log filenames.
active_learning (
ActiveLearningSimulation) – Can be used to specify an ActiveLearningSimulaiton object, which is then used to run the MD simulations while training an MTP. The calculator on the configuration is ignored in this case. Note, this does not support restarting from file.
- activeLearning()¶
- Returns:
The active learning object, if specified, otherwise None.
- Return type:
ActiveLearningSimulation | None
- addAtom(atom, md_time, time_step, kinetic_energy, incident_angle, enforce_temperature=None, label=None, post_md_hooks=None, log_interval=None, trajectory_interval=None, stability_checks=False)¶
Add an atom to the simulation.
- Parameters:
atom (PeriodicTableElement | ParticleDescriptor) – The element to add to the simulation.
md_time (PhysicalQuantity of type time) – The total MD simulation time for this event.
time_step (PhysicalQuantity of type time) – The time step of the MD simulation.
kinetic_energy (PhysicalQuantity of type energy or temperature) – The kinetic energy of the molecule when added to the system. If the kinetic energy is specified in temperature units, the translational kinetic energy will be drawn from a Maxwell-Boltzmann distribution.
incident_angle (PhysicalQuantity of type angle) – The angle between the velocity vector of the added molecule and the surface normal. An incident_angle of 0 degrees produces a molecule headed directly at the surface.
enforce_temperature (bool) – Rescale the velocities of the system to ensure the instantaneous temperature is exactly the requested temperature in the
MaxwellBoltzmannDistribution. Default: False.label (str) – A short label describing this trajectory.
post_md_hooks (list of type SPSHook) – Hook functions to call after the molecular dynamics run.
log_interval (int | PhysicalQuantity of type time) – The frequency with which log messages are written. The value can be given either as a number of MD steps (integer) or as a time interval. If not given, the default interval specified in SurfaceProcessSimulation is used.
trajectory_interval (int) – The frequency with which configurations are saved in the trajectory. The value can be given either as a number of MD steps (integer) or as a time interval. If not given, the default interval specified in SurfaceProcessSimulation is used.
stability_checks (bool) – Add check during molecular dynamics to make sure the simulation is stable and producing meaningful results. If an unstable event is encountered, the simulation will be repeated with slightly different initial conditions.
- addMolecule(molecule, md_time, time_step, kinetic_energy, incident_angle, internal_energy=None, enforce_temperature=None, label=None, post_md_hooks=None, log_interval=None, trajectory_interval=None, stability_checks=False)¶
Add a molecule to the simulation.
- Parameters:
molecule (
MoleculeConfiguration) – The molecule to add to the simulation.md_time (PhysicalQuantity of type time) – The total MD simulation time for this event.
time_step (PhysicalQuantity of type time) – The time step of the MD simulation.
kinetic_energy (PhysicalQuantity of type energy or temperature) – The kinetic energy of the molecule when added to the system. If the kinetic energy is specified in temperature units, it is treated as thermal kinetic energy and will be drawn from a
MaxwellBoltzmannDistribution. Rotation and vibration degrees of freedom are discarded.incident_angle (PhysicalQuantity of type angle) – The angle between the velocity vector of the added molecule and the surface normal. An incident_angle of 0 degrees produces a molecule headed directly at the surface.
internal_energy (PhysicalQuantity of type temperature) – The internal kinetic energy of the molecule when added to the system drawn from a
MaxwellBoltzmannDistribution. The translational kinetic energy component is replaced by the value set for the kinetic_energy parameter.enforce_temperature (bool) – Rescale the velocities of the system to ensure the instantaneous temperature is exactly the requested temperature in the
MaxwellBoltzmannDistribution. Applies to both kinetic- and internal energy. Default: False.label (str) – A short label describing this trajectory.
post_md_hooks (list of type SPSHook) – Hook functions to call after the molecular dynamics run.
log_interval (int | PhysicalQuantity of type time) – The frequency with which log messages are written. The value can be given either as a number of MD steps (integer) or as a time interval. If not given, the default interval specified in SurfaceProcessSimulation is used.
trajectory_interval (int) – The frequency with which configurations are saved in the trajectory. The value can be given either as a number of MD steps (integer) or as a time interval. If not given, the default interval specified in SurfaceProcessSimulation is used.
stability_checks (bool) – Add check during molecular dynamics to make sure the simulation is stable and producing meaningful results. If an unstable event is encountered, the simulation will be repeated with slightly different initial conditions.
- addSequence(molecule, number_of_events, md_time, time_step, mean_kinetic_energy, mean_incident_angle, std_kinetic_energy=None, std_incident_angle=None, internal_energy=None, enforce_temperature=None, relaxation_time=None, relaxation_time_step=None, fbmc_time=None, fbmc_displacement=None, optimize_geometry=None, label=None, post_md_hooks=None, log_interval=None, trajectory_interval=None, stability_checks=None)¶
Add a series of events with the given molecule or atom.
- Parameters:
molecule (PeriodicTableElement | ParticleDescriptor |
MoleculeConfiguration) – The molecule to add to the simulation. This may either be a MoleculeConfiguration or an element.
:param number_of_events:The number of cycles in which a new molecule / atom is added to the simulation. :type number_of_events:int
- Parameters:
md_time (PhysicalQuantity of type time) – The MD simulation time for the MD simulation of the initial process, where a molecule is launched at the substrate.
time_step (PhysicalQuantity of type time) – The time step of the MD simulation of the initial process, where a molecule is launched at the substrate.
mean_kinetic_energy (PhysicalQuantity of type energy or temperature) – The average kinetic energy of the molecule or atom when added to the system. If the kinetic energy is specified in temperature units, it is treated as thermal kinetic energy and will be drawn from a
MaxwellBoltzmannDistribution. For molecules, rotation and vibration degrees of freedom need to be set separately via the internal_energy parameter.mean_incident_angle (PhysicalQuantity of type angle) – The average angle between the velocity vector of the added molecule and the surface normal. An incident_angle of 0 degrees produces a molecule headed directly at the surface.
std_kinetic_energy (PhysicalQuantity of type energy) – The standard deviation of the kinetic energy. If the mean_kinetic_energy is specified in temperature units, std_kinetic_energy is automatically set to 0 since the kinetic energy will follow a
MaxwellBoltzmannDistribution.std_incident_angle (PhysicalQuantity of type angle) – The standard deviation of the incident angle.
internal_energy (PhysicalQuantity of type temperature) – The thermal kinetic energy of the molecule or atom when added to the system drawn from a
MaxwellBoltzmannDistribution. For molecules, the translational kinetic energy component is replaced by the value set for the kinetic_energy parameter.enforce_temperature (bool) – Rescale the velocities of the system to ensure the instantaneous temperature is exactly the requested temperature in the
MaxwellBoltzmannDistribution. Applies to both kinetic- and internal energy. Default: False.relaxation_time (PhysicalQuantity of type time) – The MD simulation time of an optional subsequent MD relaxation, which is run after the initial process. This additional relaxation can be run with a slightly larger time step to extend the total simulation time.
relaxation_time_step (PhysicalQuantity of type time) – The time step of an optional subsequent MD simulation, which is run after the initial process.
fbmc_time (PhysicalQuantity of type time) – The MC simulation time of an optional subsequent FBMC relaxation, which is run after the initial process. This additional relaxation can be run with a slightly larger time step to extend the total simulation time. If None, no FBMC simulation will be run.
fbmc_displacement (PhysicalQuantity of type length) – The maximum displacement of an optional subsequent FBMC simulation, which is run after the initial process.
optimize_geometry (bool) – Flag that controls if the geometry should be optimized after the FBMC run.
label (str) – A short label describing this trajectory.
post_md_hooks (list of type SPSHook | None) – Hook functions to call after each event.
log_interval (int | PhysicalQuantity of type time) – The frequency with which log messages are written. The value can be given either as a number of MD steps (integer) or as a time interval. If not given, the default interval specified in SurfaceProcessSimulation is used.
trajectory_interval (int) – The frequency with which configurations are saved in the trajectory. The value can be given either as a number of MD steps (integer) or as a time interval. If not given, the default interval specified in SurfaceProcessSimulation is used.
stability_checks (bool) – Add check during molecular dynamics to make sure the simulation is stable and producing meaningful results. If an unstable event is encountered, the simulation will be repeated with slightly different initial conditions.
- dependentStudies()¶
- Returns:
The list of dependent studies.
- Return type:
list of
Study
- directionAngles()¶
Returns the direction angle of the atoms or molecules for each event. It returns
numpy.nanfor each event that does not add atoms or molecules. It returnsNoneif this is the case for all events in the simulation.- Returns:
The direction angles.
- Return type:
PhysicalQuantity of type angle | NoneType
- filename()¶
- Returns:
The filename where the study object is stored.
- Return type:
str
- forceBiasMonteCarloEquilibration(mc_time, max_atom_displacement=None, optimize_geometry=False, log_interval=None, trajectory_interval=None)¶
Run a time-stamped force bias Monte Carlo equilibration.
- Parameters:
mc_time (PhysicalQuantity of type time) – The total elapsed Monte Carlo time to simulate.
max_atom_displacement (PhysicalQuantity of type length) – The maximum distance an atom can move in each Cartesian direction during a single step. Default: 0.1*Angstrom.
optimize_geometry (bool) – Flag controls if the geometry should be optimized after the FBMC run. Default: False.
log_interval (int | PhysicalQuantity of type time) – The frequency with which log messages are written. The value can be given either as a number of MD steps (integer) or as a time interval. If not given, the default interval specified in SurfaceProcessSimulation is used.
trajectory_interval (int) – The frequency with which configurations are saved in the trajectory. The value can be given either as a number of MD steps (integer) or as a time interval. If not given, the default interval specified in SurfaceProcessSimulation is used.
- incidentAngles()¶
Returns the incident angle of the atoms or molecules for each event. It returns
numpy.nanfor each event that does not add atoms or molecules. It returnsNoneif this is the case for all events in the simulation.- Returns:
The incident angles.
- Return type:
PhysicalQuantity of type angle | NoneType
- kineticEnergies()¶
Returns the kinetic energy of the atoms or molecules for each event. It returns
numpy.nanfor each event that does not add atoms or molecules. It returnsNoneif this is the case for all events in the simulation.- Returns:
The kinetic energies.
- Return type:
PhysicalQuantity of type energy | NoneType
- logFilenamePrefix()¶
- Returns:
The filename prefix for the logging output of the study.
- Return type:
str |
LogToStdOut
- molecularDynamicsEquilibration(md_time, time_step, post_md_hooks=None, log_interval=None, trajectory_interval=None)¶
Run an MD simulation but without adding an atom or molecule to the system.
- Parameters:
md_time (PhysicalQuantity of type time) – The total MD simulation time for this event.
time_step (PhysicalQuantity of type time) – The time step of the MD simulation.
post_md_hooks (list of type SPSHook | None) – Hook functions to call after the molecular dynamics run.
log_interval (int | PhysicalQuantity of type time) – The frequency with which log messages are written. The value can be given either as a number of MD steps (integer) or as a time interval. If not given, the default interval specified in SurfaceProcessSimulation is used.
trajectory_interval (int) – The frequency with which configurations are saved in the trajectory. The value can be given either as a number of MD steps (integer) or as a time interval. If not given, the default interval specified in SurfaceProcessSimulation is used.
- nlinfo()¶
- Returns:
Structured information about the SurfaceProcessSimulation.
- Return type:
dict
- nlprint(stream=None)¶
Print a string containing an ASCII table useful for plotting the Study object.
- Parameters:
stream (python stream) – The stream the table should be written to. Default:
NLPrintLogger()
- numberOfProcessesPerTask()¶
- Returns:
The number of processes to be used to execute each task. If None, all available processes should execute each task collaboratively.
- Return type:
int | None |
ProcessesPerNode
- numberOfProcessesPerTaskResolved()¶
- Returns:
The number of processes to be used to execute each task. Default values are resolved based on the current execution settings.
- Return type:
int
- objectId()¶
- Returns:
The name of the study object in the file.
- Return type:
str
- optimizeGeometry(max_forces=PhysicalQuantity(0.05, eV / Ang), max_steps=200, max_step_length=PhysicalQuantity(0.2, Ang), optimizer_method=None)¶
Run geometry optimization.
- Parameters:
max_forces (PhysicalQuantity of type force) – The convergence criterion for the atomic forces. Default: 0.05*eV/Angstrom.
max_steps (int) – The maximum number of optimization steps. Default: 200.
max_step_length (PhysicalQuantity of type length) – The maximum step length the optimizer may take. Default: 0.2*Ang.
optimizer_method (FIRE | LBFGS) – The optimizer to use for optimizing the structure. Default: LBFGS.
- saveToFileAfterUpdate()¶
- Returns:
Whether the study is automatically saved after it is updated.
- Return type:
bool
- sputteringYield(exclude_elements=None, start_trajectory_index=None, end_trajectory_index=None, substrate_search_radius=PhysicalQuantity(2.0, Ang))¶
Compute the average sputtering yield, that is the average number of substrate particles removed in one event. To estimate the number of events, this function only considers the ones with a label that contains ‘deposition’.
- Parameters:
exclude_elements (List of type PeriodicTableElement) – List of elements that are to be excluded from substrate atoms when computing the yield, for example the ones used for sputtering. Default: the difference between the set of elements in the final and the initial configuration.
start_trajectory_index (int) – Index of the first trajectory in the simulation, usually identifies the clean surface. Indexing based on Movie Tool. Default: the first trajectory with a ‘deposition’ label available.
end_trajectory_index (int) – Index of the last deposition trajectory in the simulation. Indexing based on Movie Tool. Default: the last trajectory with a ‘deposition’ label available.
substrate_search_radius (
PhysicalQuantityof type distance | None) – Search radius for finding substrate atoms that have become unbonded from the substrate structure. Unbound atoms that have substrate atoms above them within a cylinder of the given radius are considered to be part of the substrate structure and therefore not removed. Setting this argument to None disables this additional check. Default: 2 Angstrom.
- Returns:
The average sputtering yield.
- Return type:
float
- stickingCoefficient(exclude_elements=None, start_trajectory_index=None, end_trajectory_index=None, substrate_search_radius=PhysicalQuantity(2.0, Ang))¶
Compute average sticking coefficient, that is the average number of successful deposition events. To estimate the number of events, this function only considers the ones with a label that contains ‘deposition’.
- Parameters:
exclude_elements (List of type PeriodicTableElement) – List of elements that are to be excluded when computing the yield, for example substrate atoms or precursors that will be removed. Default: if there is any difference between the elements in the initial and final configuration, will be the elements in the initial configuration, otherwise will be an empty list.
start_trajectory_index (int) – Index of the first trajectory in the simulation, usually identifies the clean surface. Default: the first trajectory with a ‘deposition’ label available.
end_trajectory_index (int) – Index of the last deposition trajectory in the simulation. Default: the last trajectory with a ‘deposition’ label available.
substrate_search_radius (
PhysicalQuantityof type distance | None) – Search radius for finding substrate atoms that have become unbound from the substrate structure. Unbound atoms that have substrate atoms above them within a cylinder of the given radius are considered to be part of the substrate structure and therefore not removed. Setting this argument to None disables this additional check. Default: 2 Angstrom.
- Returns:
The average sticking coefficient.
- Return type:
float
- classmethod thicknessesFromSubstrate(substrate)¶
Calculate the length of the bottom fixed layer of the slab from the substrate Calculate the length of the thermostat layer of the slab (above the fixed layer) from the substrate
- Parameters:
substrate (
BulkConfiguration) – The initial surface (bulk slab model).- Returns:
The length of the bottom fixed layer of the slab. The length of the thermostat layer of the slab (above the fixed layer). The largest Z value The smallest Z value
- Return type:
tuple
- trajectories()¶
- Returns:
The list of all MD trajectories in the order they were run.
- Return type:
list of type MDTrajectory
- uniqueString()¶
Return a unique string representing the state of the object.
- update()¶
Run the calculations.
Usage Examples¶
Run an etching simulation of nitrogen on a silica surface using a ReaxFF potential.
# Attach ReaxFF calculator.
potentialSet = ReaxFF_CHONSSi_2012(strict_bondpairs = None)
calculator = TremoloXCalculator(parameters=potentialSet)
calculator.setVerletListsDelta(0.25*Angstrom)
bulk_configuration.setCalculator(calculator)
surface_process_simulation = SurfaceProcessSimulation(
substrate=bulk_configuration,
filename='sio2_n_etching.hdf5',
object_id='sps_0',
random_seed=7,
temperature=300.0*Kelvin,
fixed_thickness=4.0*Angstrom,
thermostat_thickness=8*Angstrom,
log_interval=10,
)
# Run 5 deposition events, each 20 ps long.
surface_process_simulation.addSequence(
molecule=Nitrogen,
number_of_events=5,
md_time=20*ps,
time_step=1.0*fs,
mean_kinetic_energy=25*eV,
std_kinetic_energy=0.3*eV,
mean_incident_angle=0*Degrees,
std_incident_angle=2*Degrees,
)
surface_process_simulation.update()
print('Sticking coefficient:', surface_process_simulation.stickingCoefficient())
The results from the simulation can be visualized using the Movie Tool on the QATK LabFloor.
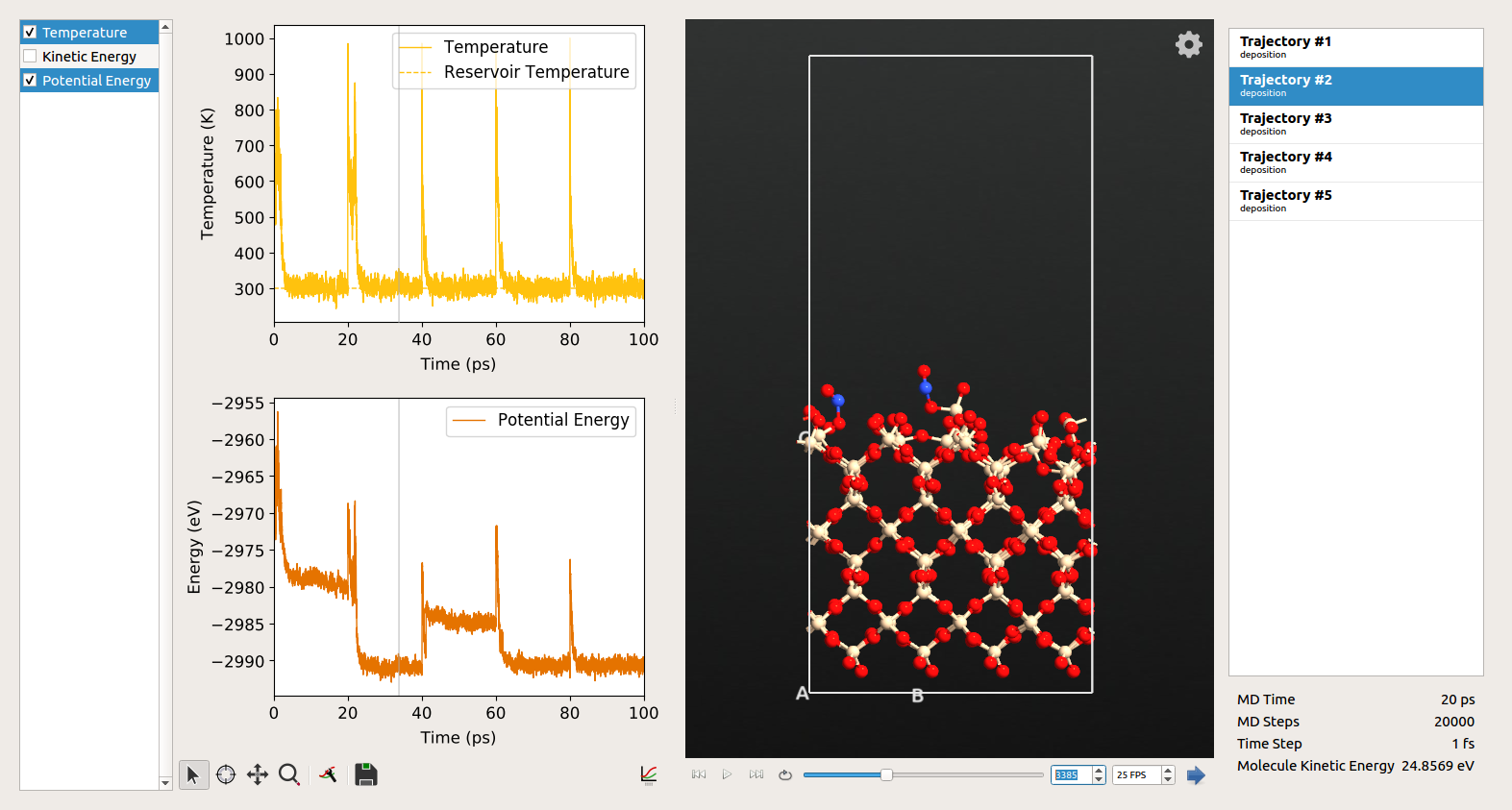
Notes¶
Note
Study objects behave differently from analysis objects. See the Study object overview for more details.
Initial setup¶
Setting up the simulation requires an initial substrate, which should be a slab
model with a surface normal in the positive z direction (such as those created
by the cleave tool available in the Builder  ).
Each simulation requires a
).
Each simulation requires a filename and a unique object_id to save the
results to, as well as a random_seed to control the random numbers used in
the simulation.
The substrate is divided into three regions, a fixed bottom layer, with
thickness controlled by fixed_thickness, a layer of thermostat controlled
atoms, with thickness controlled by thermostat_thickness, and the remaining
atoms which are unconstrained and only coupled to the thermostat indirectly
through interactions with the thermostated atoms.
Finally, the distance_above_surface parameter controls the height above the
surface that incoming molecules are placed at.
The logging and trajectory intervals can also be controlled through the
log_interval and trajectory_interval options. Both arguments take an
integer number of steps between either logging to the console or writing
trajectory data to the HDF5 file.
Adding events to the process¶
Once the object has been created, it is then possible to setup the individual
MolecularDynamics (MD) simulations. Using the addAtom or addMolecule
methods, will create a new MD simulation that adds an atom (or molecule) to the
system. The initial velocity can be controlled by the kinetic_energy and
incident_angle options. The kinetic_energy determines the magnitude of
the velocity (all atoms in the molecule will be given the same velocity
vector). While incident_angle controls the direction. The
incident_angle is defined relative to the surface normal, so a value of 0
degrees is straight into the surface and an angle of 90 degrees is parallel to
the surface.
Often, the time step must be quite small to accurately model the dynamics of
high energy particles, but can be increased after a short time to equilibrate
the system. This can be achieved by calling molecularDynamicsEquilibration
after addAtom (or addMolecule). This will add a MD simulation that does
not add any atoms, but allows for the selection of a different time step.
In addition to MD, it is also possible to run ForceBiasMonteCarlo
(fbMC) simulations using the forceBiasMonteCarloEquilibration method.
Computing the yield of a process¶
It is possible to compute the average yield of a sputtering, etching or
deposition process with the sputteringYield() and
stickingCoefficient() functions.
In order to compute the average yield, the number of events is needed, this is
estimated from the number of events with a label that contains deposition.
The default labels for addSequence(), addAtom() and addMolecule()
meet this requirement.
The optional parameters start_trajectory_index and end_trajectory_index
expect the same indexing convention used in the Movie Tool.
Usually, some atoms in the system are not supposed to be considered for the
yield estimate, this can be set through the exclude_elements, sensible
default values should cover the most common use cases.
Attention
In order to estimate the yield of the process, it is essential to have saved trajectories that correctly represent the initial and the final configuration. Make sure that (at least) the start and the end trajectories have more than one saved step, this can be tuned for each event with the trajectory_interval parameter.
SurfaceProcessSimulation Hooks¶
Hooks functions can be used to apply some changes on the last step of an event. After a sputtering event, it may be useful to clean the vacuum region from sputtered atoms with CleanVacuumRegion, or to remove the atoms of the element used for the process with RemoveElementFromSubstrate. In some cases the thermalization of the substrate could require several steps, it may then be convenient to decrease the kinetic energy of all the particles in one step with ThermalizeSubstrate.