Li-air battery interface¶
Version: 2015.1
Li-ion batteries are, at the moment, the most widely used in most most electric vehicles and hybrid electric vehicles. However, there are some disadvantages such as high price, slow charging, and low energy/power density. Li-air batteries are attracting attention due to the higher energy storing capacity and are being seen as possible alternative for Li-ion batteries. Nevertheless, still a lot of research has to be done in order to make Li-air battery competitive. In particular, various complex chemical and electrochemical side-reactions occurs at the interfaces. Li2O2/Li2CO3 is indeed a very important interface since Li2CO3 is formed at the cathode with Li2O2 when carbonate-based electrolytes are used. In this tutorial you will use QuantumATK to create the Li2O2/Li2CO3 and use ATK study its electronic properties. You will compare the results with Ref. [1].
You will:
Import/create Li2O2 and Li2CO3 bulk structures.
Cleave surfaces.
Create Li2O2/Li2CO3 interface.
Create device and optimize geometry.
Calculate electronic properties of the Li2O2/Li2CO3.
Li2O2 bulk and surface structures¶
Bulk structure¶
Open the Builder and use to add the Li2O2 structure to the Stash.
Tip
You can use the plugin to get the corresponding space group, for example

Optimize the lattice parameters by running the
Li2O2_opt.pyscript or use the experimental ones if you wish. In this tutorial you will use the GGA-RBE exchange-correlation potential by following the computational details as in Ref. [1].
Cleave surface¶
Use the tool to cleave the Li2O2 (0001) surface from the optimized Li2O2 structure as illustrated in the next figures.


Electronic structure at zero bias¶
In the Li2O2_opt.py script a few analysis have been added such as the 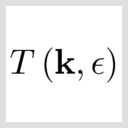 TransmissionSpectrum analysis which you can find loaded in the LabFloor once your simulation is done.
TransmissionSpectrum analysis which you can find loaded in the LabFloor once your simulation is done.

From the figure above, you can see that Li2O2 shows a wide band gap and no electronic conduction is expected at low bias voltages. Moreover, as demonstrated in [1], when introducing Li vacancies the electronic conduction decreases even more.
Note
You can follow the Transmission spectrum of perfect sheets of graphene and MoS2 tutorial for more details and how to set up a transmission spectrum analysis for a perfect periodic bulk system.
Li2CO3 bulk and surface structures¶
Bulk structure¶
The Li2CO3 bulk structure is not present in the internal database of QuantumATK.
You need to get the structure file, usually a CIF file, from an external database, e.g. you can use the Crystallography Open Database.
Search, download and copy the 9008283.cif file to your project folder.
It will be automatically loaded in the LabFloor.
For more options see Import XYZ, CIF, CAR, VASP Files in QuantumATK tutorial.

When interfacing the Li2CO3 surface with the Li2O2 you will need to use a large cell to get a small lattice mismatch. This is because the CO3 planes in the C2/c space group are slightly misaligned from the cell axes. In this tutorial you will use a modified Li2CO3 structure with space group Cm where the CO3 planes are perpendicular to the (011) plane. Also, this is the structure used in cite:Yedilfana2015.
Download the Li2CO3.py script and import the structure to the Builder to analyze it.

Cleave surface¶
Use the tool to cleave the (011) surface as indicated in the figures below.


The Li2O2/Li2CO3 interface¶
Open the tool and drag and drop the Li2CO3 surface and the optimized Li2O2 surface to the first and second drop zones.
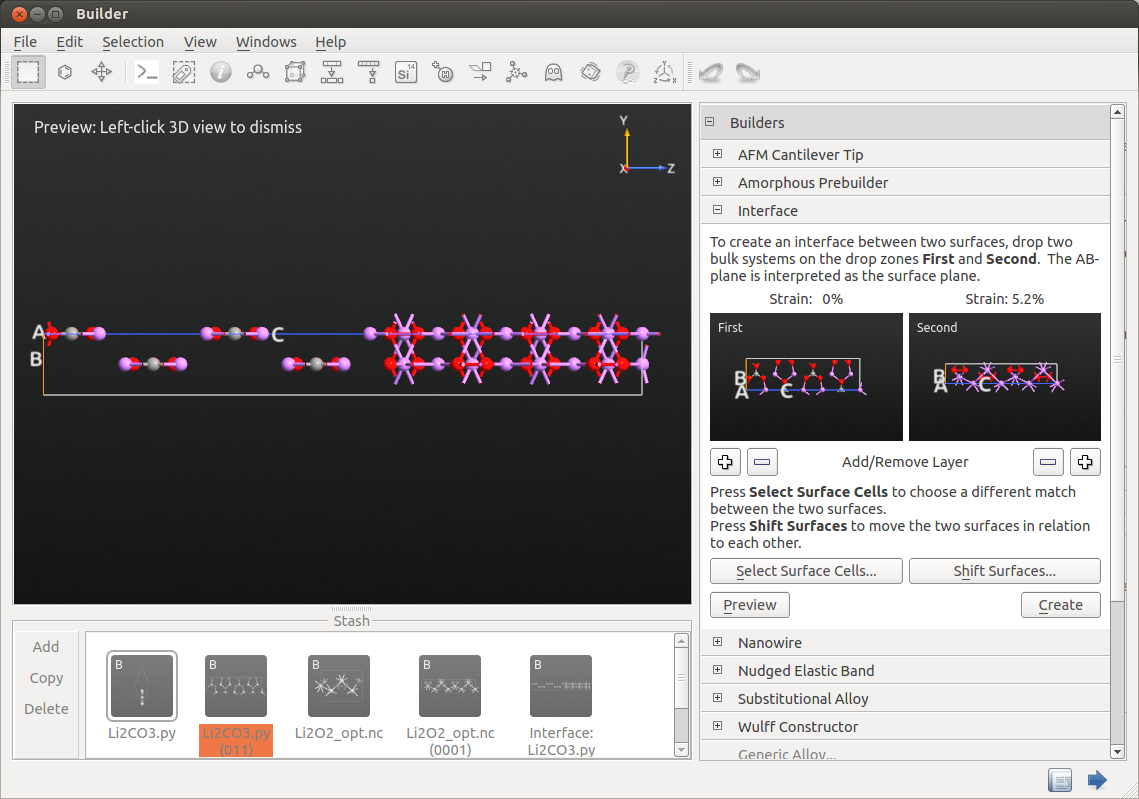
The default results are correct. However, if you want to choose another interface you can click on Select Surface Cells button and choose the desired matching parameters.

You can now create the device configuration with the plugin. The default electrode lengths are fine.

You can download this structure here: device_configuration_initial.py.
Note that in this final structure the Li2CO3 side has been slightly shifted towards the Li2O2 side to allow for a proper relaxation
Tip
You can learn more about the Interface Builder in the Technical Notes on Interface Builder.
Optimizing the interface¶
A step-by-step procedure on how to optimize the geometry of a device configuration is described in the Advanced device relaxation - manual workflow. The main steps are the following:
Tag the electrode extension regions by using the plugin. You can do that by selecting the atoms in the electrodes, the electrode extension region will be automatically selected.

Hint
Tag also the first layer outside the electrode extension regions which is already part of the central region.By doing so, the Device From Bulk plugin will be able to recognize the proper periodicity and recreate the device structure (see below).
Extract the central region as BulkConfiguration by using the
 split device plugin, increase the C vector and center the configuration.
split device plugin, increase the C vector and center the configuration.
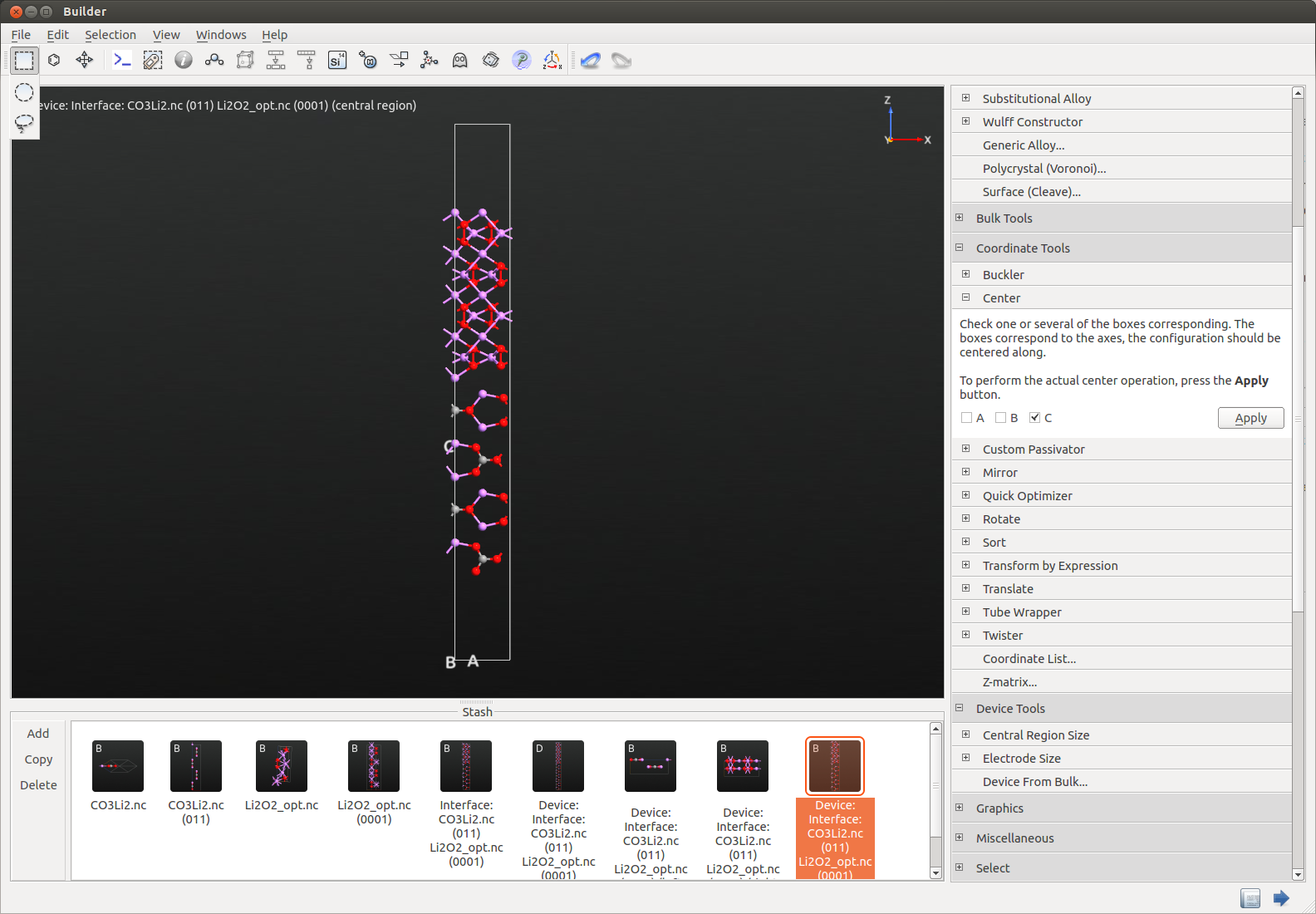
Set up a geometry optimization calculation with rigid body constrain for the Li2O2 tagged region and fixed constrain for the Li2CO3 one. You can also download the corresponding
interface_bulk_opt.py.Finally, when the optimization of the interface in a bulk configuration is done, import in the Builder the optimized structure and recreate the device as described in the previous section.
Electronic structure¶
Send the device configuration from the Stash to the Scripter and set up a new device calculator with the GGA-RPBE exchange-correlation potential and a 7x15x200 k-point sampling.
You can leave all other parameters as defaults. Here, you can download the whole script: device_interface.py.
You can add various analysis to investigate in more detail the electronic structure and the transport properties. Below, for example, the projected device density of state analysis is shown in the picture. Here you can see that Li2CO3, on the left side, has a much higher band gap than Li2O2. Electronic conduction is thus expected to be very low, as it is indeed shown in [1].

Hint
You can further investigate the Li diffusion processes in Li2O2 and Li2CO3 crystals by following the Li-ion diffusion in LiFePO4 for battery applications.
References¶