Geometry optimization: CO/Pd(100)¶
Version: 2015.1
In this tutorial you will learn how to do a geometry optimization using Fixed and Rigid constrains on the atoms. As an example, you will study adsorption of the CO molecule on a Pd(100) surface. You will use the ATK-DFT calculator engine and a standard GGA functional.
Specifically, you will:
optimize the bulk palladium crystal;
cleave the bulk to create the Pd(100) surface and relax it;
adsorb the CO molecule in the surface, and relax the system in two steps;
first apply Rigid constraints on atoms during an initial relaxation;
then apply Fixed constraints on the bottom Pd atoms in a final optimization.
lastly, relax the CO molecule and calculate the adsorption energy.

Bulk palladium¶
Create a new project called “Pd100_CO” and open the Builder  .
.
Use to import the Pd bulk to the Stash and send the configuration to the Script Generator
 using the send to button
using the send to button  .
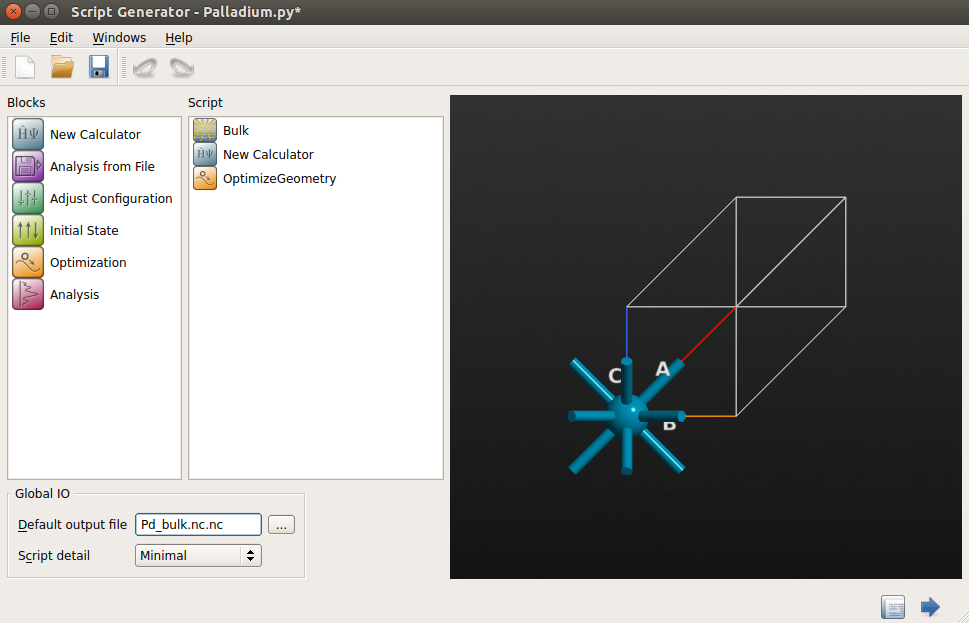
.In the Script Generator, add the
 New Calculator and
New Calculator and
 OptimizeGeometry blocks to the script by double-clicking
them, and change the default output file name to
OptimizeGeometry blocks to the script by double-clicking
them, and change the default output file name to Pd_bulk.hdf5.
Double-click the
New Calculator block and set the following
parameters in the window that opens up:k-point sampling: 9x9x9;
exchange-correlation: GGA-PBE functional.
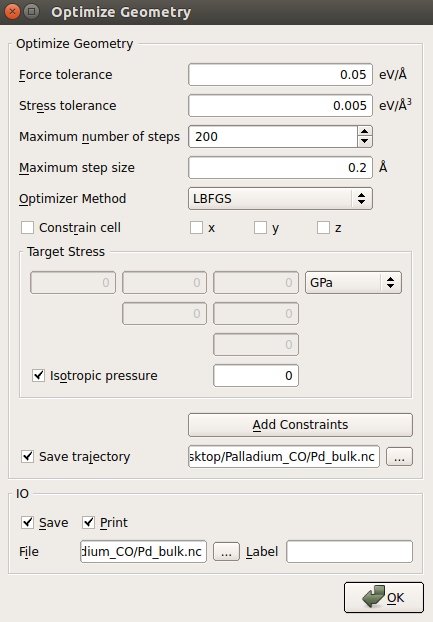
Open the
OptimizeGeometry block, and set the parameters
as shown in the image below. In particular:enable stress relaxation by unticking Constrain cell;
decrease the stress tolerance to 0.005 eV/Å3;
save the relaxation trajectory by ticking Save trajectory and enter
Pd_bulk.hdf5for the trajectory file name.
Tip
It is usually a good idea to save the trajectory file. In case your geometry optimization is interrupted you can simply restart it from one of the latest images in the trajectory. If you would like to know more about how to restart a stopped calculation, check out the tutorial Restarting a stopped calculation.
Click OK and the OptimizeGeometry window will close.
The script is now finished. Send it to the Job Manager
 using the button. Save the script in the window that appears and
run the calculation. It will take a few seconds. If needed, you can download
the script here:
using the button. Save the script in the window that appears and
run the calculation. It will take a few seconds. If needed, you can download
the script here: Pd_bulk.py.
Once the bulk relaxation is finished, the calculation output will appear on the LabFloor. The bulk configurations with IDs glD000 and glD002 are the initial and final structures, respectively, while the relaxation trajectory has ID glD001.
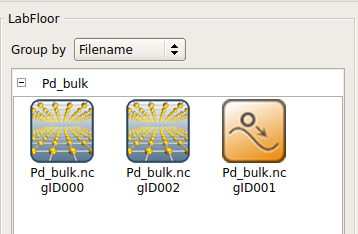
You can drag and drop these objects to the Viewer  to visualize
them. In the Viewer, you can check the lattice constant by hovering the mouse on
the cell boundaries. You could also check the lattice constant in the newly formed
log file.
to visualize
them. In the Viewer, you can check the lattice constant by hovering the mouse on
the cell boundaries. You could also check the lattice constant in the newly formed
log file.
In the following, you should of course use the relaxed bulk structure, glD002.
Build the Pd(100) surface and relax it¶
Drag and drop the relaxed bulk configuration to the Builder
 .
.Open the plug-in.
Keep the default Miller indices and click Next.
Keep the default 1x1 surface lattice and click Next.
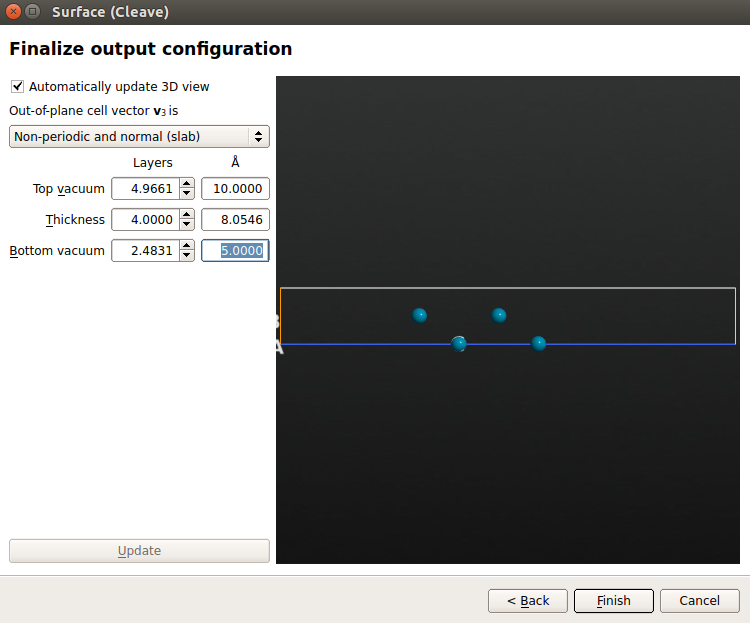
Choose a Non-periodic and normal (slab) out-of-plane cell vector, 4 slab metal layers, and 10 Å and 5 Å for the top and bottom vacuums, respectively. Click Finish to build the surface.
Send the slab structure to the Script Generator
and add the New Calculator,
OptimizeGeometry and  TotalEnergy blocks to the script,
and set the default output file name to
TotalEnergy blocks to the script,
and set the default output file name to Pd100.hdf5.
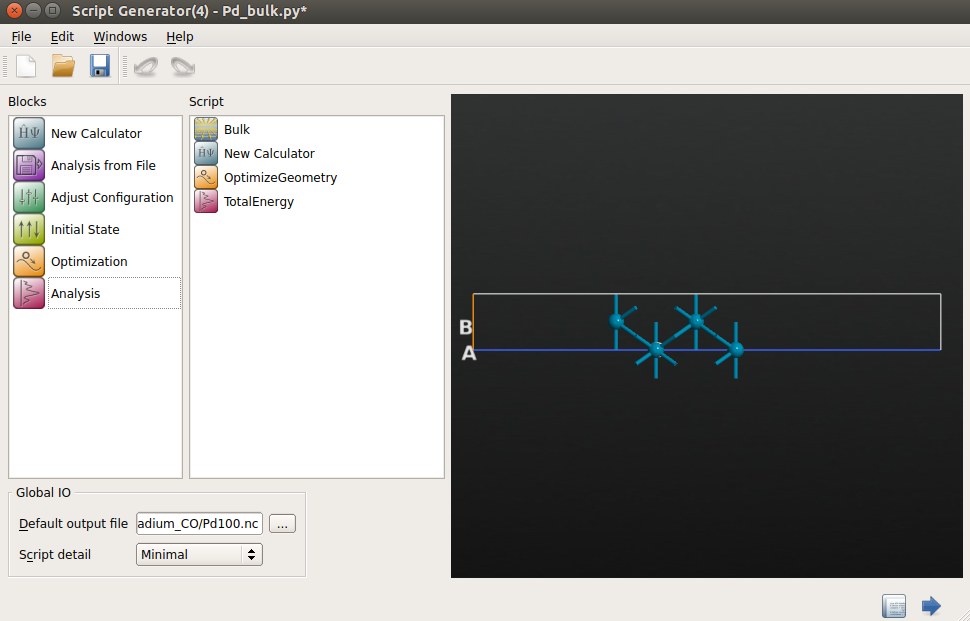
Choose the following set of calculator parameters:
k-point sampling: 9x9x1 k-points;
exchange-correlation: GGA-PBE;
FFT2D Poisson solver with Neumann boundary conditions under the slab and Dirichlet boundary conditions above the slab:
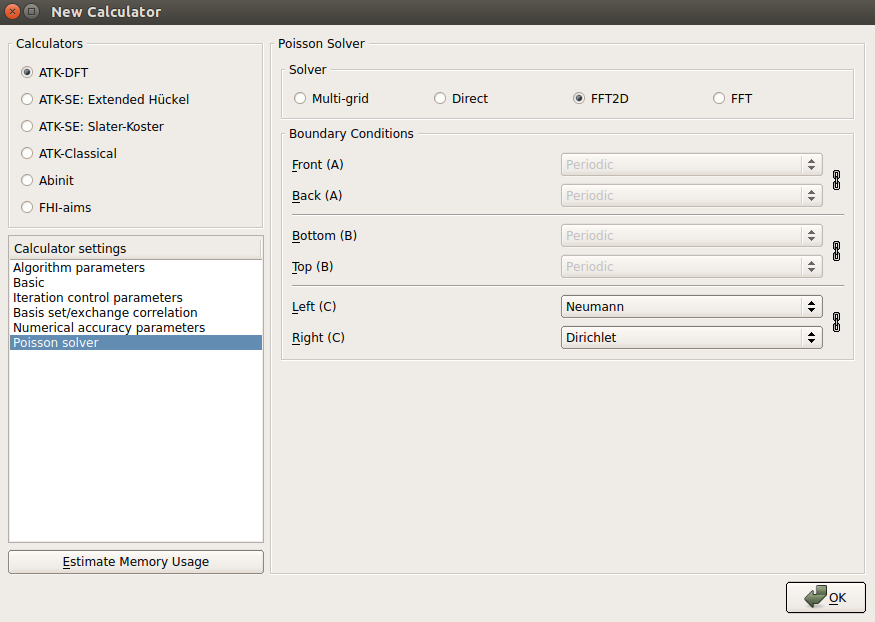
Note
This particular set of boundary conditions along the C direction of the simulation cell is appropriate for slab calculations, where the vacuum above the slab could in principle extend to infinity.
Open the
OptimizeGeometry block and set it up
for force relaxation:save the trajectory to
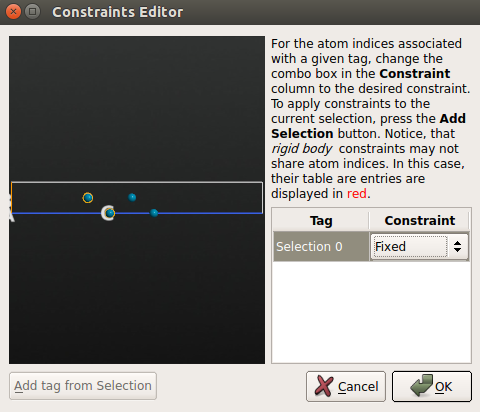
Pd100_traj.hdf5;click Add Constraints to open the Constraints Editor;
use the mouse to select the bottom two layers of the Pd(100) surface and click Add tag from selection. Those two atoms are now tagged “Selection 0”. Then choose the Fixed constraint for the “Selection 0” group.
There is nothing to edit in the
TotalEnergy block,
so the script is now finished. Save it as Pd100.pyand execute it using the Job Manager. The calculation will
take roughly one minute. If needed, you can download the script here:
Pd100.py.
Once the Pd(100) surface has been geometry optimized, the results will
appear on the LabFloor. You can again visualize the relaxation
trajectory using the Viewer .
Tip
You can check the total energy of the relaxed Pd(100) surface by selecting the TotalEnergy object and clicking on the Text Representation plugin. The energy is also printed in the log file.
Relax the CO/Pd(100) system¶
The next step is to add the CO molecule to the surface and relax the system.
You should use the CO/Pd(100) structure available here: CO_Pd100.py. If needed,
you can learn how to add a molecule to a surface from the tutorial Building molecule–surface systems: Benzene on Au(111).
As mentioned in the introduction, you will relax the CO/Pd(100) configuration in two steps:
use a Rigid constraint for CO and Fixed constrain for the surface, together with a relatively low electron density mesh cut-off, in order to get a quick idea of where the CO will be adsorbed;
the final relaxation step does not constrain the CO molecule and just fixes the bottom of the Pd(100) slab.
Rigid relaxation¶
Transfer the provided CO/Pd(100) configuration (CO_Pd100.py) to the
Script Generator and add the New Calculator
and OptimizeGeometry blocks. Enter
Pd100_CO_rigid.hdf5 for the default output file name, and then
edit the script:
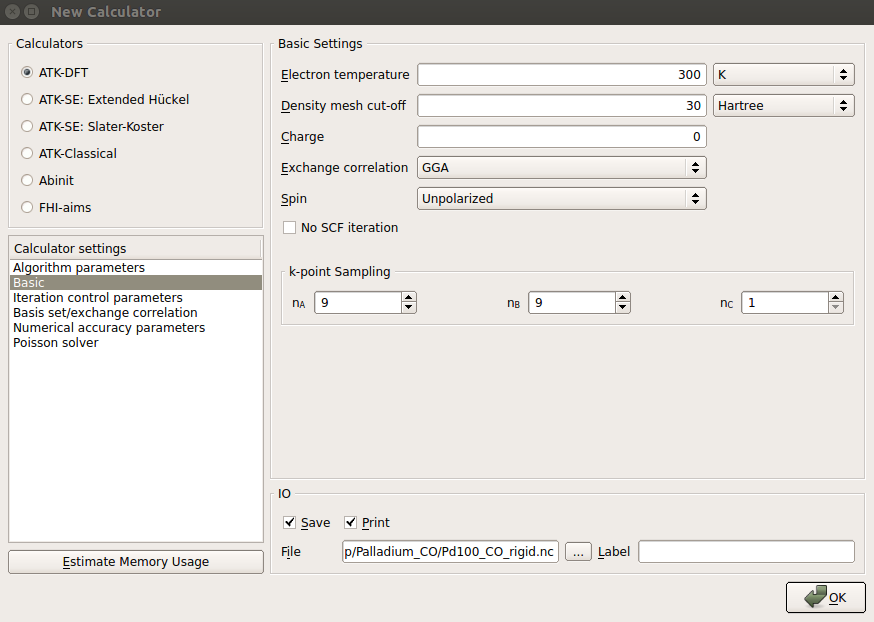
In the
New Calculator block, use the same
settings as for the Pd(100) relaxation in the previous section,
but decrease the density mesh-cut off to 30 Hartree.
Set up the
OptimizeGeometry block similarly
to the one used for Pd(100), but choose a slightly different set of
constraints:select the Fixed constraint for all atoms in the metal slab;
select the Rigid constraint for the CO molecule (add the C and O atoms to the group “Selection 1”).
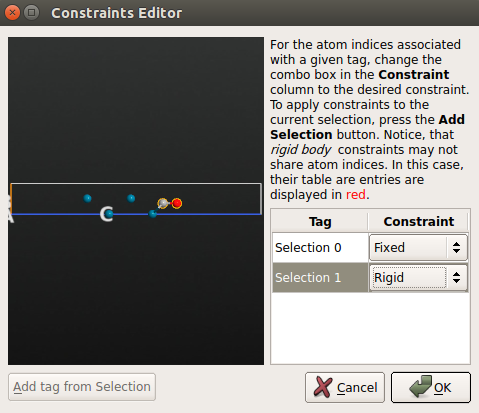
Note
The Rigid constraint on a group of atoms causes those atoms to move like a rigid body during force optimization.
Save the script as
Pd100_CO_rigid.pyand run it using the Job Manager. The simulation should take roughly 5 minutes if executed in serial. You can download the script here:Pd100_CO_rigid.py.
Important
If you take a look at the trajectory, you will see that the CO molecule has moved slightly away from its initial position, but the C–O bond length has not changed, and the molecule has not rotated. This is how the rigid constraint works.
Final relaxation¶
You will now perform the final geometry optimization of the CO/Pd(100) structure, starting from the result of the previous step.
Send the configuration with ID glD002 in
Pd_CO_rigid.hdf5to the Scripter, and enterPd_CO.hdf5for the default output file name.Once again, add the
New Calculation,
OptimizeGeometry and TotalEnergy blocks, and edit them
similarly to what you did in the previous section. This time, however, use a
75 Hartree mesh-cutoff for the ATK-DFT calculator, and use only a Fixed
constraint for the bottom two Pd atoms:
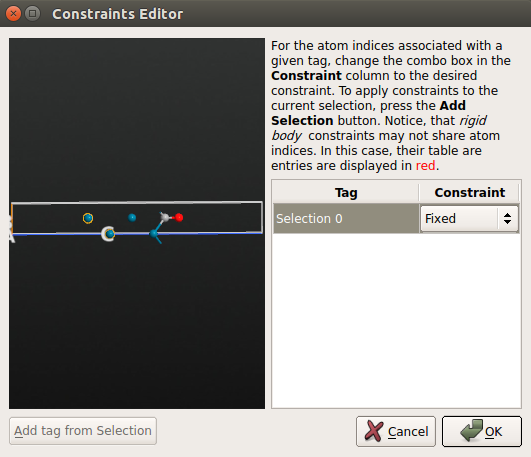
Save the script as
Pd_CO.pyand execute it using the Job Manager or in a Terminal. The calculations should take about 20 min in total, but can of course be quicker if run in parallel on more CPUs. The script is also available here:Pd100_CO.py.
Tip
You can actually use the Viewer tool to monitor the progress
of the geometry optimization while the calculation is running. The trajectory
object is continuously updated as new relaxations steps are taken.
When the calculation has finished you should have a look at the full relaxation trajectory, and see how the CO molecule has moved from a position around the hollow adsorption site on the Pd(100) surface to a bridge site between two Pd surface atoms:
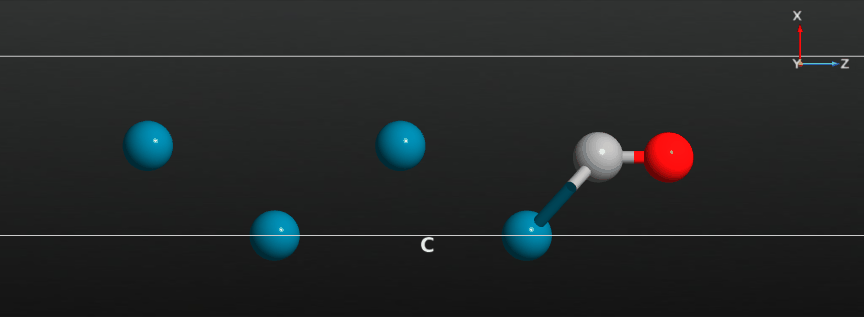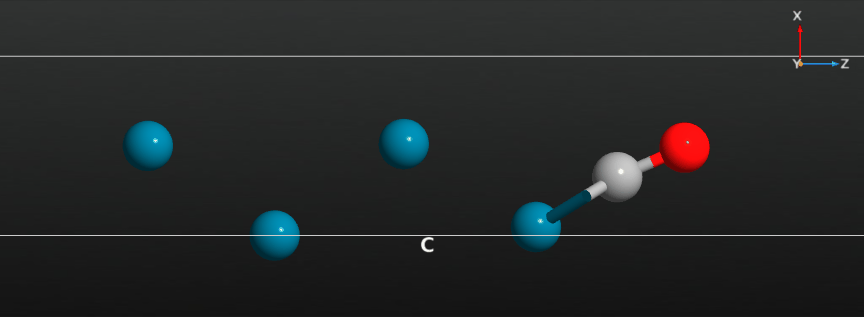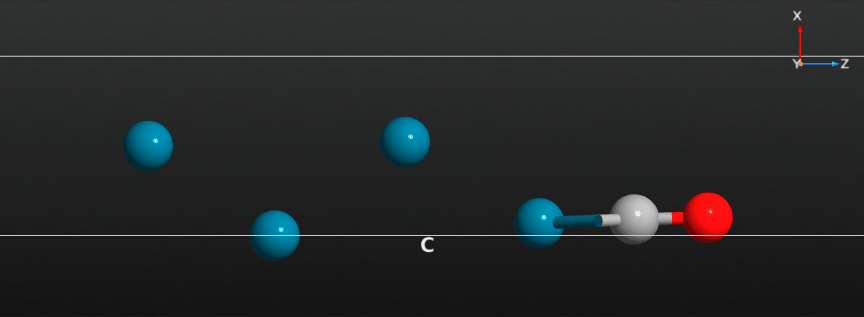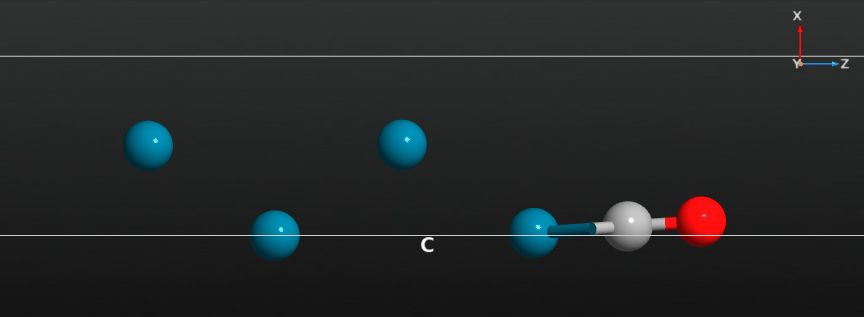
Relax the CO molecule¶
In order to calculate the CO adsorption energy on Pd(100) you also need to relax the isolated CO molecule. We can obtain it following the next steps:
go to the Builder and add a new configuration using ;
click the Molecular Builder tool and click in the panel that opens up;
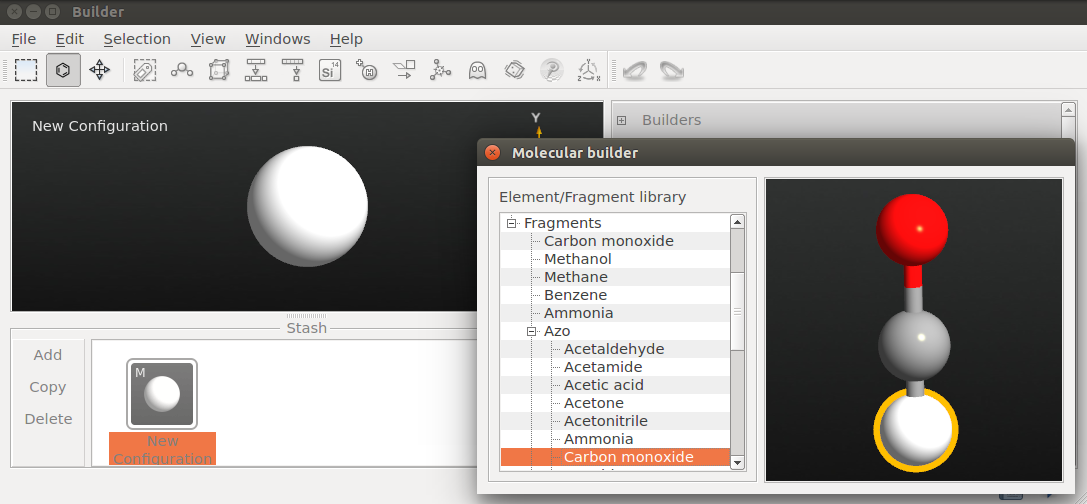
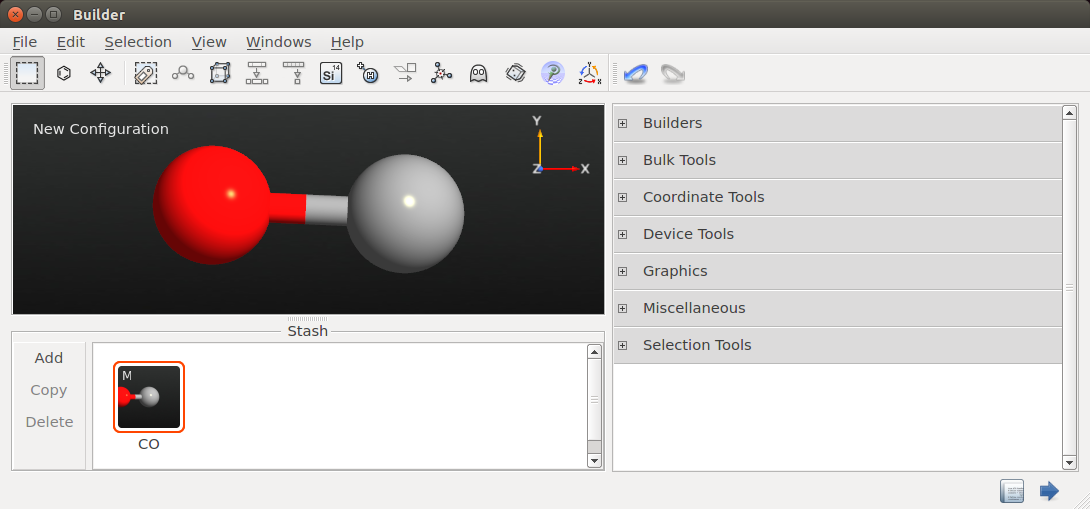
next, click the hydrogen atom in the
New ConfigurationStash item to substitute it for the CO fragment, close the Molecular Builder window, and rename the Stash item toCO.
Tip
For more details about how to use the Molecular Builder, you can check out this tutorial: Molecular builder.
Now you just need to relax the CO atomic coordinates and compute the total energy.
Send the structure to the Script Generator and add the
New Calculator, OptimizeGeometry and
TotalEnergy blocks. Enter CO.hdf5 for the default output file name,
and then edit the blocks a bit:
- New Calculator: Choose the GGA-PBE functional;
- OptimizeGeometry: You may want to save the
trajectory or decrease the force tolerance, but it’s not mandatory;
Save the script as CO.py and run the calculation – it should
be very fast. The script is also available for download: CO.py.
You will find that the C–O bond length relaxes to 1.143 Å, which matches experiments very well.
Adsorption energy¶
You are now ready to calculate the CO/Pd(100) adsorption energy at a full monolayer coverage, \(\Delta E\), which is of course given by total energy differences:
You can use the Text Representation plugin to see all three total
energies and then calculate \(\Delta E\), or you can use a script to do it: adsorption_energy.py.
The adsorption energy, calculated using PBE and the FHI pseudopotentials with a DZP basis set, is \(\Delta E\) = –1.97 eV. However, this number is affected by the so-called basis set superposition error, so you should proceed to the next section to correct for this.
Counterpoise correction¶
The basis set superposition error (BSSE) is due to the incompleteness of the LCAO basis set, and can have a significant impact on energy differences between different sub-systems. As explained in a tutorial (DFT-D and basis-set superposition error), the superposition of Pd and CO basis sets in the CO/Pd(100) system can give rise to an artificial lowering of the total energy because the Pd(100) and CO sub-systems can “borrow” basis functions from each other. The result is a too large adsorption energy.
The standard approach to neutralize the BSSE is to apply a so-called
counterpoise correction. In QuantumATK, this is done using the
counterpoiseCorrected.
You should therefore calculate the counterpoise (CP) corrected total energy for the CO/Pd(100) system in order to compute the CP corrected adsorption energy:
Use the script shown below (can be downloadad here: Pd_CO_cp.py). The script reads
in the previously relaxed CO/Pd(100) configuration and the calculator
that was used, and creates a new calculator that applies the CP correction.
The configuration is then relaxed and the total energy is calculated.
Run the script using the Job Manager or in a Terminal. It should perform 4 very small relaxation steps, and finish within 10 minutes. The CP correction will increase the total energy of the CO/Pd(100) system, resulting in an adsorption energy of \(\Delta E^\mathrm{CP}\) = –1.68 eV.
1# -------------------------------------------------------------
2# Bulk Configuration
3# -------------------------------------------------------------
4
5bulk_configuration = nlread('Pd100_CO.nc', BulkConfiguration)[-1]
6
7# Add tags
8bulk_configuration.addTags('CO', [4, 5])
9bulk_configuration.addTags('Pd', [0, 1, 2, 3])
10bulk_configuration.addTags('Selection 0', [0, 1])
11
12# -------------------------------------------------------------
13# Calculator
14# -------------------------------------------------------------
15
16# Get the calculator settings from non-BSSE calculation.
17calculator = bulk_configuration.calculator()
18basis_set = calculator.basisSet()
19exchange_correlation = calculator.exchangeCorrelation()
20numerical_accuracy_parameters = calculator.numericalAccuracyParameters()
21poisson_solver = calculator.poissonSolver()
22
23# Create BSSE calculator.
24bsse_calculator = counterpoiseCorrected(LCAOCalculator, ["CO", "Pd"])
25calculator = bsse_calculator(
26 basis_set=basis_set,
27 exchange_correlation=exchange_correlation,
28 numerical_accuracy_parameters=numerical_accuracy_parameters,
29 poisson_solver=poisson_solver,
30 )
31
32bulk_configuration.setCalculator(calculator)
33nlprint(bulk_configuration)
34bulk_configuration.update()
35nlsave('Pd100_CO_cp.nc', bulk_configuration)
36
37# -------------------------------------------------------------
38# Optimize Geometry
39# -------------------------------------------------------------
40constraints = [0, 1]
41
42bulk_configuration = OptimizeGeometry(
43 bulk_configuration,
44 max_forces=0.05*eV/Ang,
45 max_steps=200,
46 max_step_length=0.2*Ang,
47 constraints=constraints,
48 trajectory_filename='Pd100_CO_cp.nc',
49 disable_stress=True,
50 optimizer_method=LBFGS(),
51 )
52nlsave('Pd100_CO_cp.nc', bulk_configuration)
53nlprint(bulk_configuration)
54
55# -------------------------------------------------------------
56# Total Energy
57# -------------------------------------------------------------
58total_energy = TotalEnergy(bulk_configuration)
59nlsave('Pd100_CO_cp.nc', total_energy)
60nlprint(total_energy)