To import a SiO2 (quartz) structure from the NanoLab’s Internal Database, follow the step-by-step protocol described below.
In the BuilderStash (1), click on the Add button (2) and choose From Database (3) to open NanoLab’s Crystal Database.
Use the Search bar (1) to find SiO2 (quartz) structure (2) and click on the Add button (3) to add the structure to the BuilderStash.
Tip
Internal NanoLab structure library contains several hundred basic molecule, crystal, fullerene, zeolite, monomer and 2D material structures. If you cannot find there a structure of your interest, click on Databases tool to use NanoLab’s interface to query online databases such as Crystallography Online Database and Materials Project.
To create a workflow for OptimizeGeometry calculations in the Workflow Builder, follow the step-by-step protocol described below. Here, we will use DFT with LCAOCalculator basis sets and a hybrid HSE06 DFT functional, as it is optimized for bulk semiconductor and insulator materials.
In the Builder, use the Send content to other tools button to send the imported SiO2 (quartz) structure to the Workflow Builder.
Go to the Block Catalog ‣ QuantumATK tab ‣ Calculators category on the right hand panel and drag-and-drop the SetLCAOCalculator block to the middle Workflow Area ‣ Build tab.
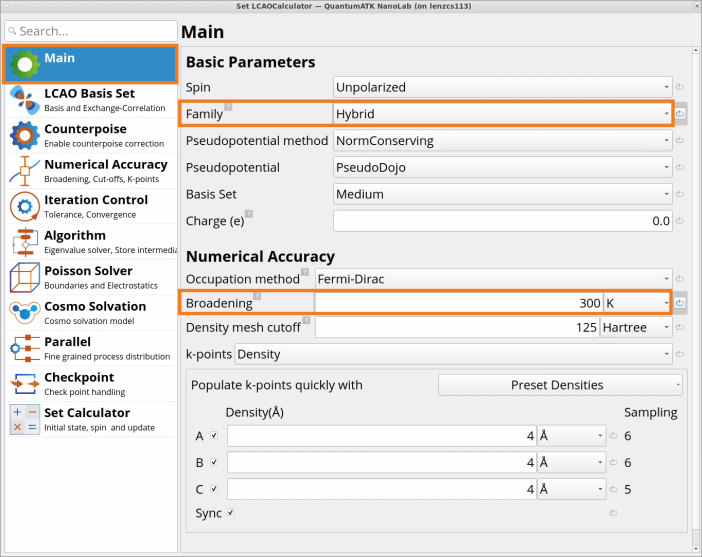
Double-click on the SetLCAOCalculator block and edit the Main tab:
Change the Exchange CorrelationFamily from the default GGA to Hybrid with a predefined default HSEO6 (HybridGGA.HSE06) functional (functional can be changed under the LCAO Basis Set tab).
Change the numerical accuracy paremeter Broadening from 1000 K to 300 K (recommended for semiconductor materials).
Tip
SetLCAOCalculator block sets medium k-point sampling, medium basis set and 125 Ha density mesh cutoff (system-dependent) by default, typically providing good accuracy results with modest computational resources. It is recommended though to check the convergence of your results with these parameters.
SetLCAOCalculator block uses a default Fermi-Dirac Occupation Method with 1000 K Broadening, which generally works reasonably well across various systems. Another setting may be more appropriate for your system, and you can adjust these settings according to the recommendations in Occupation Methods for semiconductor, insulator, metal, and molecule systems.
For slabs, surfaces and interfaces (modelled as slabs), it is recommended to set FFT2D Poisson Solver (Dirichlet/Neumann) in the out-of-plane (C) direction instead of the default FFT (Periodic) in the SetLCAOCalculator block, Poisson Solver/Solver Type section. For neutral molecules, stick to the default FFT Poisson solver, whereas for charged molecules, set the Multigrid/Multipole Poisson Solver. Read these technical notes on Poisson solvers to learn more.
Go to the Block Catalog ‣ QuantumATK tab ‣ Optimization and Dynamics category on the right hand panel and drag-and-drop the OptimizeGeometry block to the middle Workflow Area ‣ Build tab.
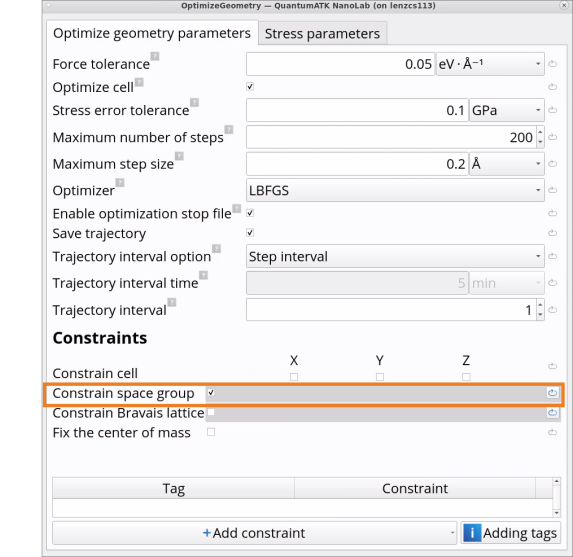
Double-click on the OptimizeGeometry block and change from Constrain Bravais Lattice to Constrain space group.
Change the Workflow name in the Workflow Stash and Filename as needed (e.g., SiO2-geom-opt-HSE06.hdf5).
Click on the Send content to other tools button and choose Jobs as a script to send the workflow to the Jobs tool in order to submit this job on a local machine or a remote cluster. It is recommended to parallelize over both MPI processes and threads in DFT simulations. Read the technical notes on Parallelization of QuantumATK calculations to learn more.
This also generates a Python script SiO2-geom-opt-HSE06.py, based on this workflow. It can be opened and edited with the Editor tool and then sent to the Jobs tool.
Tip
Constrain space group relaxes atom positions, unit cell volume and shape and preserves crystal symmetry during the geometry optimization, whereas the default Constrain Bravais Lattice allows to relax to a different crystal symmetry. Constrain Bravais lattice can also be used when optimizing the geometry of alloys, amorphous materials or crystals with defects with a preserved Bravais lattice type.
Unit cell volume and shape can be constrained by unchecking the box Optimize cell in the OptimizeGeometry block, thus allowing for local relaxation of atoms only.
When doing geometry optimization of a bare surface, or, e.g., surface covered by molecules, it is possible to Add constraints on selected (tagged) atoms or monolayers in the OptimizeGeometry block to, e.g., fix the bottom substrate layer.
In case of calculating PhononBandstructure, VibrationalMode, DielectricTensor (or other properties that require DynamicalMatrix calculations), it is recommended to pre-optimize structure with a tighter force (e.g., 0.001) and stress (e.g., 0.01) tolerances than default.
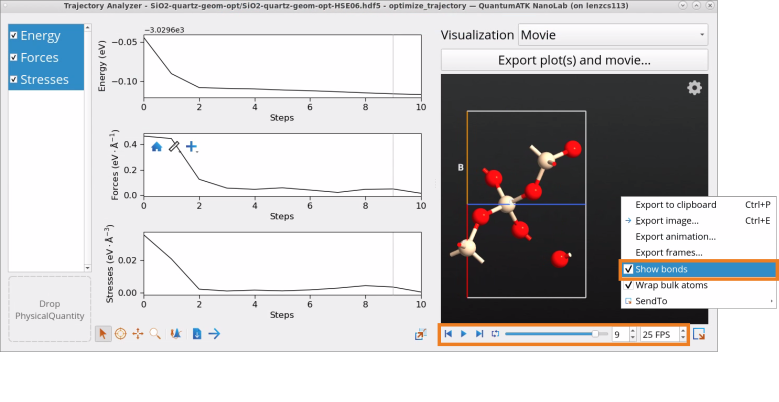
Once the calculation has finished, we are ready to analyze the results.
Go to the Data View tool.
In the Data Sources select the file SiO2-geom-opt-HSE06.hdf5 and then in the Data View window select both, BulkConfiguration_0 (initial geometry) and optgeom (optimized geometry), followed by the right-click to Open All with Text Representation.
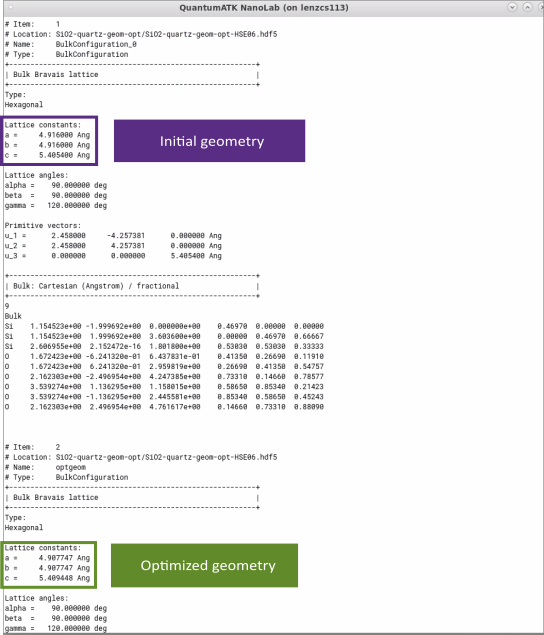
Inspect and compare lattice constants of initial and optimized geometries in text representation. The optimized lattice constants of SiO2 (quartz) , a=4.908 Å and c= 5.409 Å, with the hybrid HSE06 functional are in a good agreement with experimental results a=4.913 Å and c=5.405 Å [1].
Right-click on the optimize_trajectory object in the Data View window and open it with the Movie Tool to investigate the convergence of energy, forces and stresses throughout the simulation. It took 10 steps to converge energy, forces and stresses to the default force and stress tolerance values.
Right-click on the structure in the Movie tool to check the box Show bonds and use the Play button to see the evolution of structure throughout the geometry optimization simulation.
Note that lattice parameters optimized with the default, computationally-cheaper GGA PBE functional instead, a=4.966 Å and c=5.466 Å, deviate by ~1% from the measured values, compared to ~0.1% when optimized with the more sophisticated hybrid HSE06 functional used in this tutorial example for SiO2 (quartz).

 Builder Stash (1), click on the
Builder Stash (1), click on the  Add button (2) and choose From Database (3) to open NanoLab’s Crystal Database.
Add button (2) and choose From Database (3) to open NanoLab’s Crystal Database.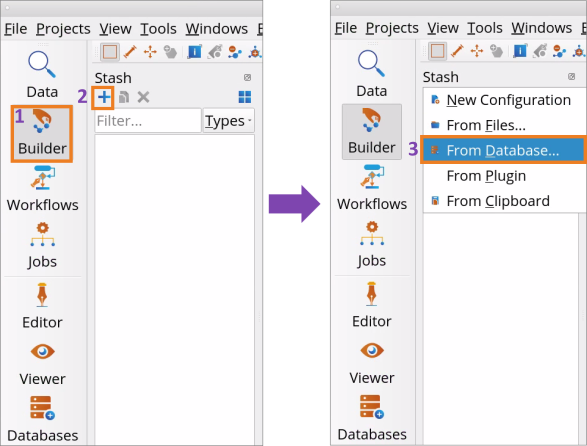
 Search bar (1) to find SiO2 (quartz) structure (2) and click on the
Search bar (1) to find SiO2 (quartz) structure (2) and click on the 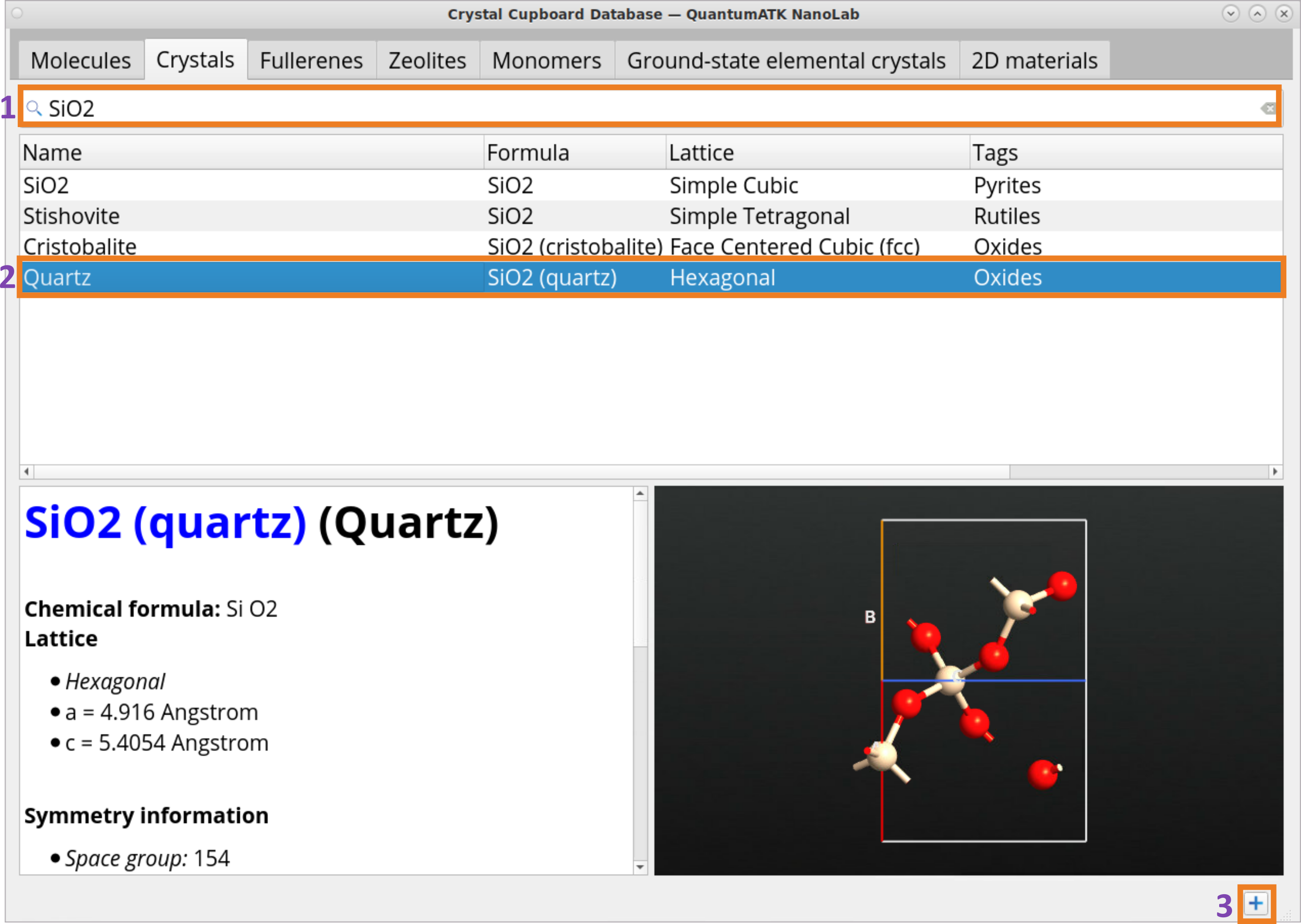
 Databases tool to use NanoLab’s interface to query online databases such as Crystallography Online Database and Materials Project.
Databases tool to use NanoLab’s interface to query online databases such as Crystallography Online Database and Materials Project. Workflow Builder, follow the step-by-step protocol described below. Here, we will use DFT with LCAOCalculator basis sets and a hybrid HSE06 DFT functional, as it is optimized for bulk semiconductor and insulator materials.
Workflow Builder, follow the step-by-step protocol described below. Here, we will use DFT with LCAOCalculator basis sets and a hybrid HSE06 DFT functional, as it is optimized for bulk semiconductor and insulator materials. Send content to other tools button to send the imported SiO2 (quartz) structure to the
Send content to other tools button to send the imported SiO2 (quartz) structure to the 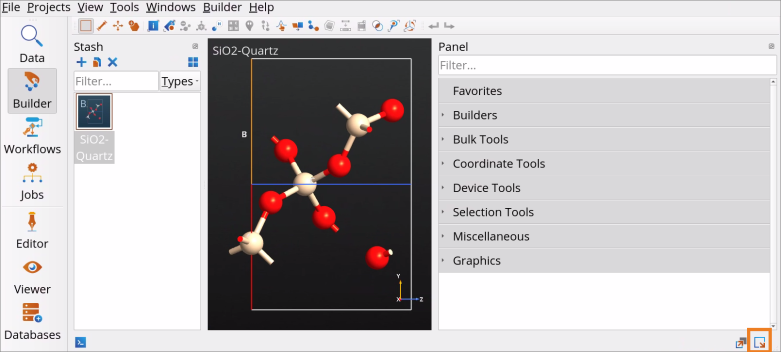
 SetLCAOCalculator block to the middle Workflow Area ‣ Build tab.
SetLCAOCalculator block to the middle Workflow Area ‣ Build tab.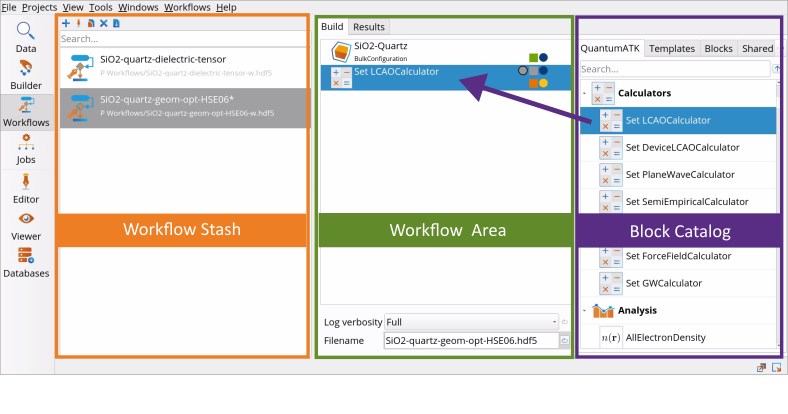
 OptimizeGeometry block to the middle Workflow Area ‣ Build tab.
OptimizeGeometry block to the middle Workflow Area ‣ Build tab.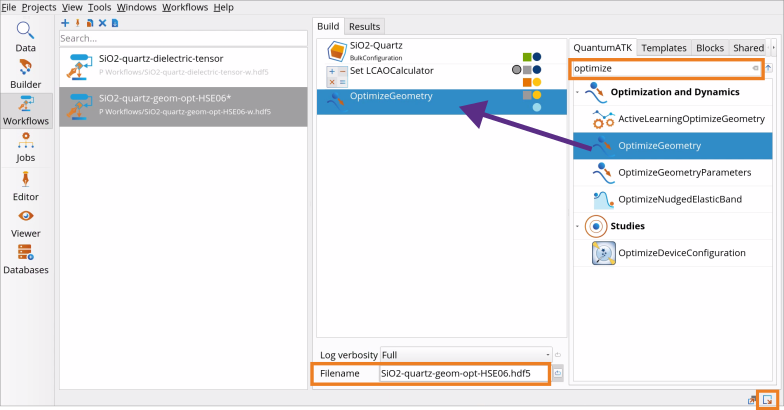
 Jobs tool in order to submit this job on a local machine or a remote cluster. It is recommended to parallelize over both MPI processes and threads in DFT simulations. Read the technical notes on Parallelization of QuantumATK calculations to learn more.
Jobs tool in order to submit this job on a local machine or a remote cluster. It is recommended to parallelize over both MPI processes and threads in DFT simulations. Read the technical notes on Parallelization of QuantumATK calculations to learn more. Editor tool and then sent to the
Editor tool and then sent to the  BulkConfiguration_0 (initial geometry) and
BulkConfiguration_0 (initial geometry) and 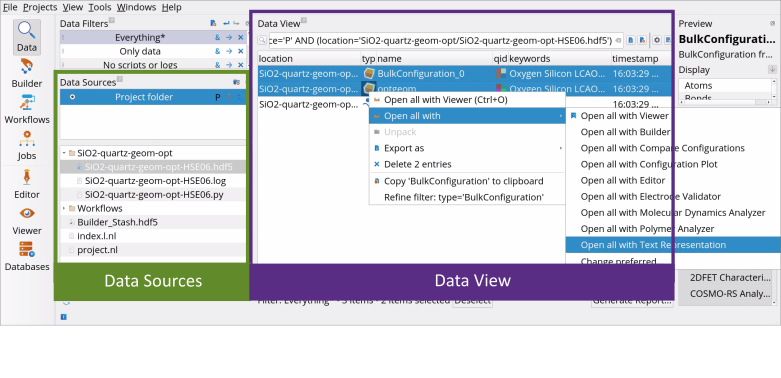
 Play button to see the evolution of structure throughout the geometry optimization simulation.
Play button to see the evolution of structure throughout the geometry optimization simulation.