Ammonia inversion reaction barrier using DFTB and NEB¶
In this tutorial, you learn how to use a Nudged Elestic Band to calculate the reaction path of ammonia inversion. You will use the Density Functional based Tight Binding (DFTB) method (dftb.org) in order to make calculations fast.
Setting up the NEB object¶
Open the
 Builder and click .
Builder and click .The Database tool will open. From the menu, select . Type “Ammonia” in the search field, and you should see the following:

Double-click the “Ammonia” entry (or click the
 icon), and the ammonia molecule will be added to the Stash.
icon), and the ammonia molecule will be added to the Stash.
Note
When running NEB calculations, it is important to restrict at least a few degrees freedom. What primarily happens in the ammonia reaction considered here, is that the nitrogen atom moves through the hydrogen plane. It is therefore a good idea to keep the hydrogen atom plane fixed when defining the initial reaction path. This can be acheived rather easily, see blow.
In the Builder, activate the Move Tool
 .
Select each of the hydrogen atoms as anchor atoms (marked by a red halo); this is the atom you will operate on.
Since no atoms were selected when the Move Tool was opened,
all atoms will simultaneously be selected (marked by a yellow halo); these atoms will be affected by any operation carried out in the Move Tool.
.
Select each of the hydrogen atoms as anchor atoms (marked by a red halo); this is the atom you will operate on.
Since no atoms were selected when the Move Tool was opened,
all atoms will simultaneously be selected (marked by a yellow halo); these atoms will be affected by any operation carried out in the Move Tool.In the Move widget, enter the value 0 for the z coordinate of the atoms, and press Enter.

Close the Move tool by closing the widget or clicking the Select tool.
Now ensure that the ammonia molecule is selected on the stash by left-clicking it. Then click the Copy button . This adds an additional, identical ammonia molecule to the Stash.
Open the Mirror tool from the Coordinate Tools in the right tool bar. Ensure that the mirror plane has a normal parallel to the Z axis and contains the origin (this is the default), and press Apply to invert the molecule.

You have now defined the initial and final states of the reaction path. The next step is to set up the NEB configuration by inserting a number of images in-between these states.
To this end, open the Nudged Elastic Band tool under Builders from the plugin panel, and drag the two configurations into the Initial and Final slots.
Set the maximum distance between images to 0.09 Å in order to have an odd number of images. This is very important, since the reaction path is symmetric. Choose the Linear method and press Create. The NEB object will now be built and added to the Stash.
Push the Play button to see the different configurations that will be used as starting configurations when optimizing the reaction pathway.
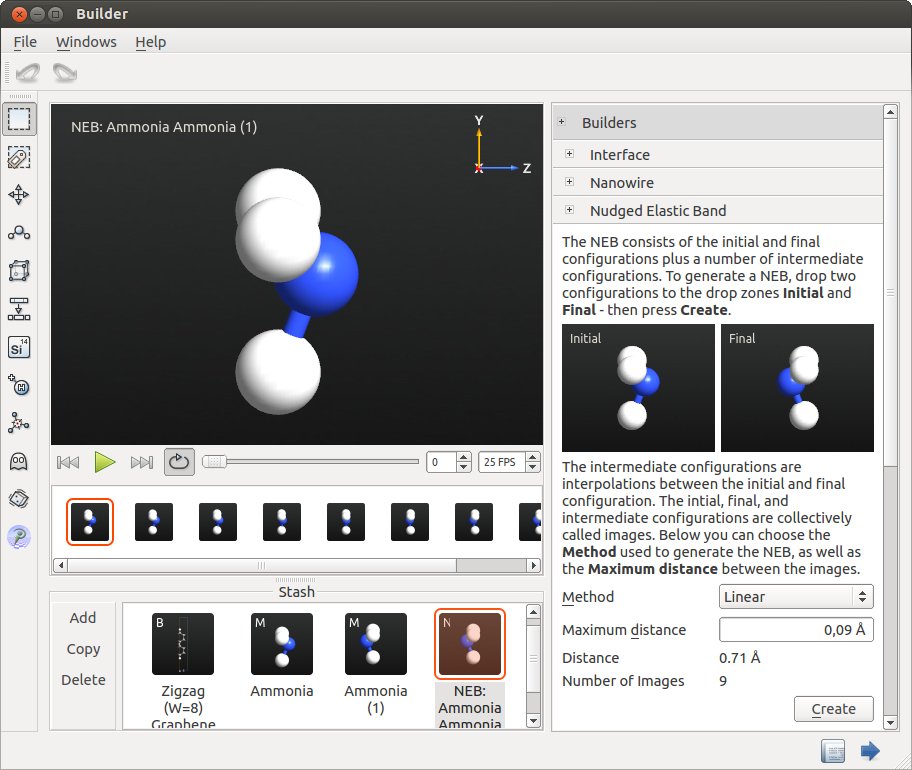
Performing the NEB simulation¶
To set up the actual calculations, send the NEB object to the  Script Generator using the Send To button
Script Generator using the Send To button  .
.
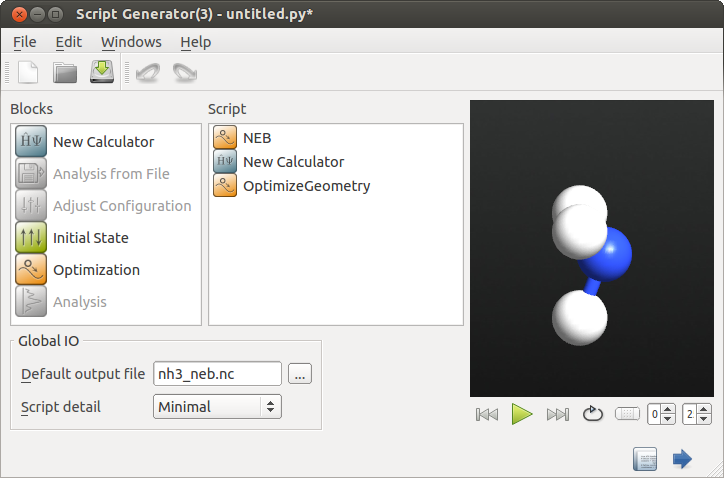
In the Script Generator, add the following blocks:
 New Calculator
New Calculator OptimizeGeometry
OptimizeGeometry
and change the default output name to nh3_neb.nc.
Open the New Calculator block and edit it:
Select the ATK-SE: Slater-Koster calculator.
Go to the Slater-Koster basis set and check that the DFTB [mio] basis set is selected.
Uncheck the No SCF iteration box.

Open the OptimizeGeometry block, and make sure it looks as shown below.

You are now ready to perform the NEB optimization. Send the script to the  Job Manager and execute it.
Job Manager and execute it.
The optimization will take 20-30 minutes depending on your computer.
Note
While most other implementations of the DFTB model use a pointwise electrostatic pair potential interaction model, ATK-SE uses a Poisson solver for calculating the electrostatic interactions. This approach is significantly slower for small systems, like the current ammonia molecule, but faster for large systems, where it scales as \(O(N)\) instead of a typical \(O(N^2)\) scaling.
The use of a Poisson solver ensures that the DFTB model is equivalent to the other calculators in QuantumATK, and thus can be used for modeling device geometries and allows for using implicit solvent models.
Analyzing the NEB simulation¶
Return to the main QuantumATK window, and select the file nh3_neb.nc.
Select the last NEB configuration in the file (the one with the highest ID number).
Press the Show configuration and you will see the calculated energy barrier and the corresponding configurations in the reaction path.
The reaction path can also be illustrated as an animation by pressing the Play button.

Fig. 121 Reaction path of ammonia inversion, computed with the DFTB [mio] method. The end-points were not optimized.¶
Attention
The obtained reaction barrier is not fully accurate, since the end points were not optimized. This is apparent from the fact that the second image has a lower energy than the end-points. For real studies with the NEB method, the end-points should always be optimized first.
Tip
You can closely inspect an individual optimized configuration in the reaction path by clicking the dots in the reaction paths plot (and stopping the movie).
A recipe for faster calculations¶
In the calculation above, you used a linear interpolation between the end-points to set up the initial guess for the NEB path. In many cases the linear path is, however, very far from the optimized one. Comparing the movie of the converged result above to the initial NEB path you set up, it can be seen that that the hydrogen atoms need to move out-wards to make room for the nitrogen atom as it passes through the center of the molecule, and then they move back in.
If this behavior could somehow be part of the initial path, one would save some steps in the ionic dynamics, and hence computational time. However, since the initial and final positions of the hydorgen atoms are identical, this cannot be captured by the linear interpolation.
There are, however, other methods for generating the initial path, and some are available in ATK.
Go back to the
Builder, and the Nudged Elastic Band plugin.Keep the image distance, but change the Method to Image Dependent Pair Potential, and create the NEB.
If you now inspect the initial path, you can see that it already contains this behavior of the hydrogen atoms. This is one of the strengths of the IDPP method [1] for NEB calculations.
Follow the same procedure as above to run the NEB calculation via the
Scripter,
and you will find that it runs about 30% faster this time, but that results are identical.
Note
The IDPP method is similar to the Halgren–Lipscomb method [2], which is also available in ATK (but not suitable for this particular system).
References