Electronic Properties of Phase Change Material Ge2Sb2Te5¶
Version: T-2022.03
In this tutorial, we will investigate electronic properties of Ge2Sb2Te5, a phase change material used in non-volatile memory technology, using Density Functional Theory (DFT) calculations. Please follow the tutorial Band Structure of a SiC Crystal to familiarize yourself with the use of scripter to set up calculations. Only the final scripts will be provided in this tutorial.

Geometry¶
For this tutorial, let us first download the unitcell configuration of Ge2Sb2Te5, in a trigonal crystal system with hexagonal lattice symmetry, from the materials project database (mp-1224375) using the Database Tool available in the NanoLab.
For reference, the geometry can also be downloaded from here GST-geometry.py.
Bandgap Calculation¶
As the first property, let us compute the bandgap of Ge2Sb2Te5 and compare to literature data. To do that, the unit cell geometry will be optimized with space group constraint for minimum energy first and then its BandStructure will be computed. Additionally, we will compare the default GGA (PBE XC functional) results to more accurate HSE06 calculations using the PseudoDojo LCAO basis sets. To be exact, 4 different calculation settings will be used as follows:
PBE functional based geometry relaxation and BandStructure calculation(
PBE.py)HSE06 hybrid functional based geometry relaxation and BandStructure calculation(
HSE.py)HSE06 hybrid functional with ADMM basis set for both geometry relaxation and BandStructure calculation(
HSE-ADMM.py)PBE functional based geometry relaxation followed by HSE06 hybrid XC functional based BandStructure calculation(
PBE-HSE.py)
The bandgap can be quickly viewed from the BandStructure object preview window or from the BandStructure Analyzer object. The corresponding results of the above calculations (using 40 MPI processes in 1 node) are given in the below image.
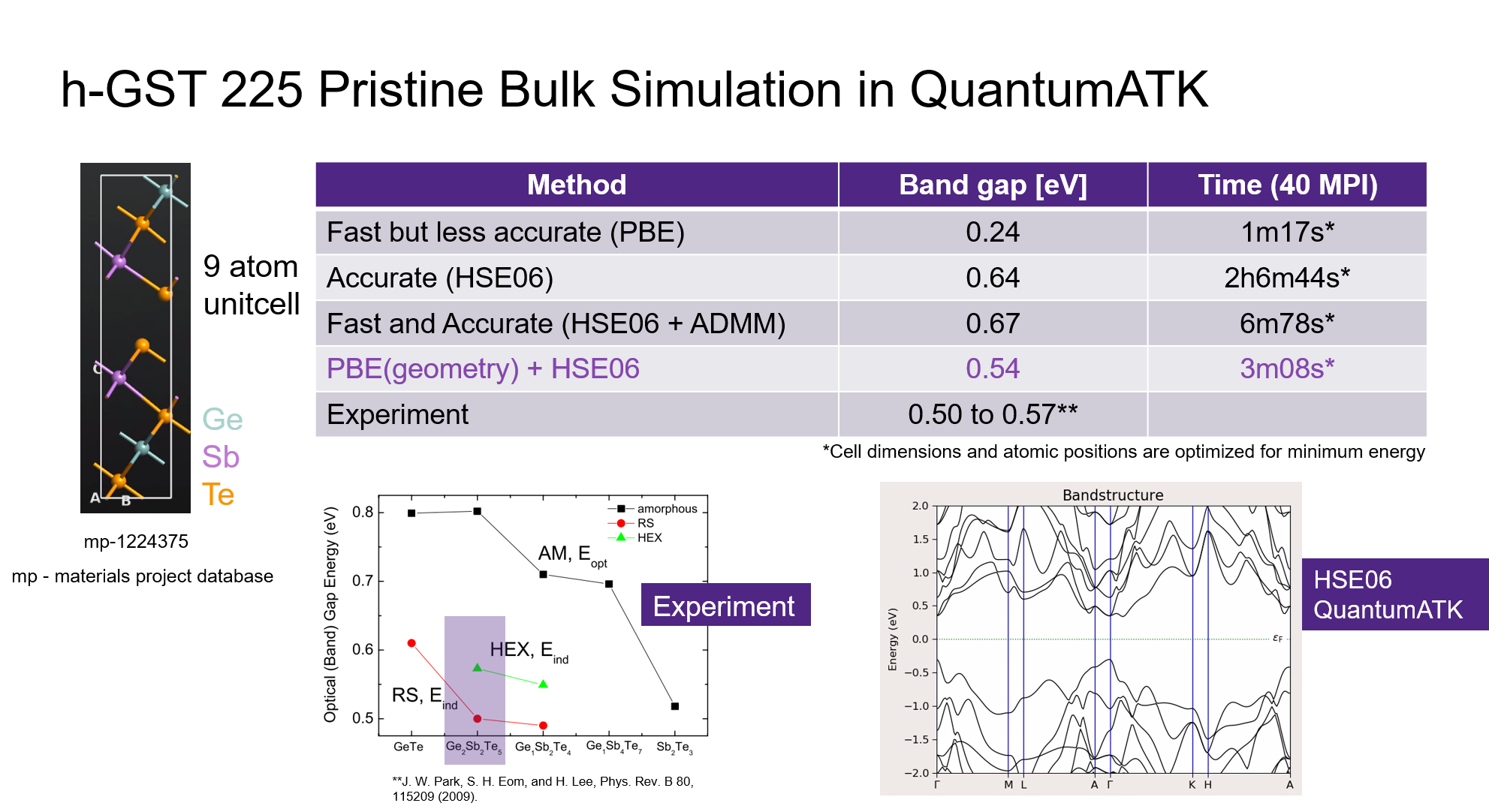
Experimental bandgap of hexagonal Ge2Sb2Te5 is known to be between 0.50 and 0.57 eV [1]. PBE only calculation is the fastest but the bandgap is underestimated to 0.24 eV only. Standard HSE06 calculation took over 2 hours and gave a bandgap of 0.64 eV. Inclusion of ADMM basis sets accelerated the HSE06 calculation to completion within 7 minutes and produced a similar bandgap. However, HSE06 based bandstructure calculation on PBE optimized geometry gave a better bandgap value of 0.54 eV while taking only about 3 minutes.
Lattice Parameters¶
Lattice parameters of the Ge2Sb2Te5 unitcell after geometry optimization using the PBE and HSE06 functionals are given in the below image.
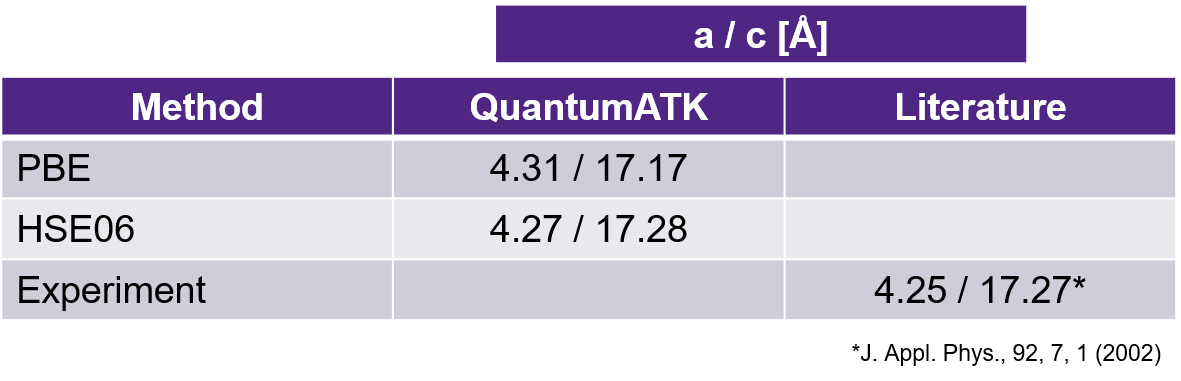
Both PBE and HSE06 optimized geometries have lattice parameters within 0.1 Å of the experimental values [2].
Cohesive Energies¶
Cohesive energies of the Ge2Sb2Te5 is computed using PBE and HSE06 methods using Ebulk - (2*EGe + 2*ESb + 5*ETe) and the results are shown below. Here, Ebulk is the total energy of bulk system and EGe, ESb and ETe are atomic reference energies of Ge, Sb and Te, respectively.
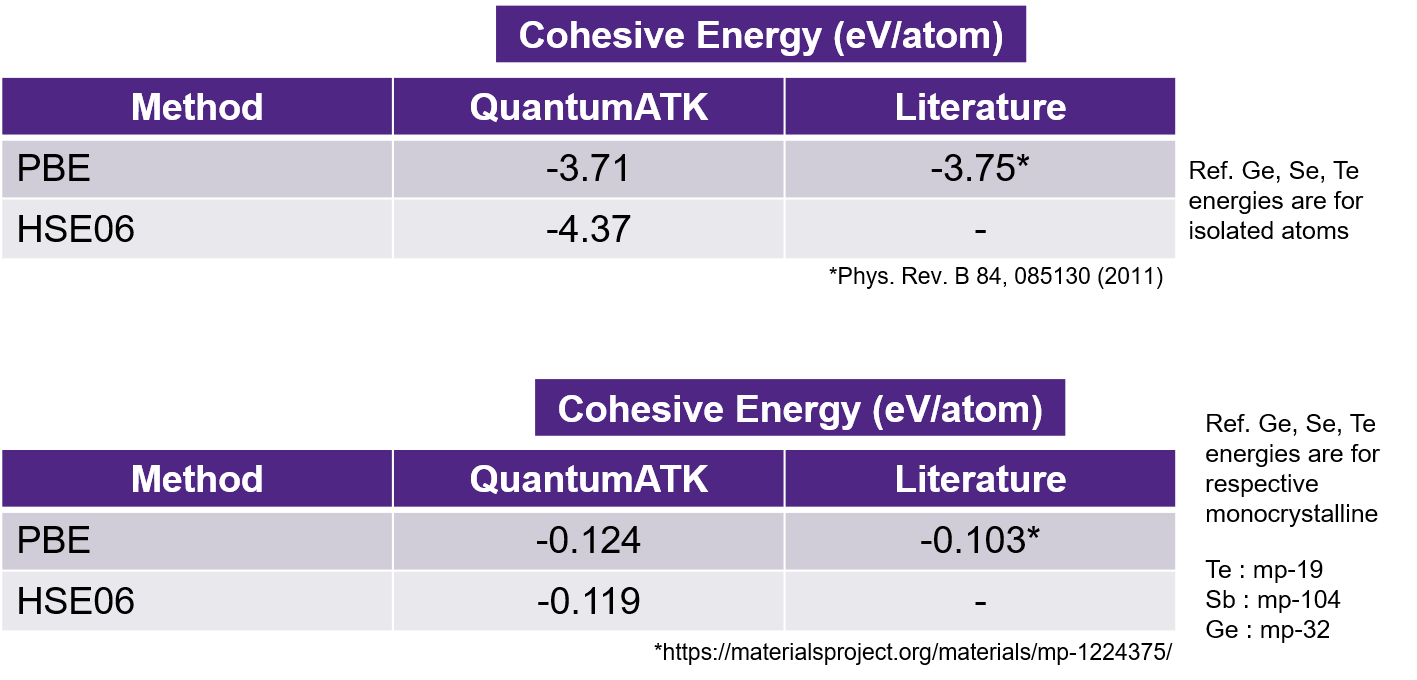
In the above results, two different sets of reference energy values are used for the elemental contributions.
First set of cohesive energies (top of the picture) are computed using total energies of isolated Ge, Sb and Te atoms as reference and match well with literature [3].
The following scripts are used for the above reference calculations (Ge (HSE, PBE), Sb (HSE, PBE), Te (HSE, PBE)).
For the second set, total energies per atom in homogeneous bulk systems are used. The corresponding scripts are attached here (Ge, Sb, Te). Reference cohesive energy is calculated using the data from materials project database using the following IDs (GST:mp-1224375, Ge:mp-32, Sb:mp-104, Te:mp-19).
From the above image, we find that our calculations agree reasonably with the published calculated reference values within 4 meV/atom for isolated atom reference and within 20 meV/atom for bulk reference. Note that the literature reference uses a different basis set and calculator settings.
Neutral Vacancy Formation Energies¶
In this section, we compute the formation energies of neutral vacancies within a unitcell of Ge2Sb2Te5 using PBE XC functional and compare to literature results [4]. The formation energy of a single neutral vacancy is obtained as Epristine GST - (EGST with vacancy + Ebulk reference), where Ebulk reference is the energy of the removed atom in its bulk state.
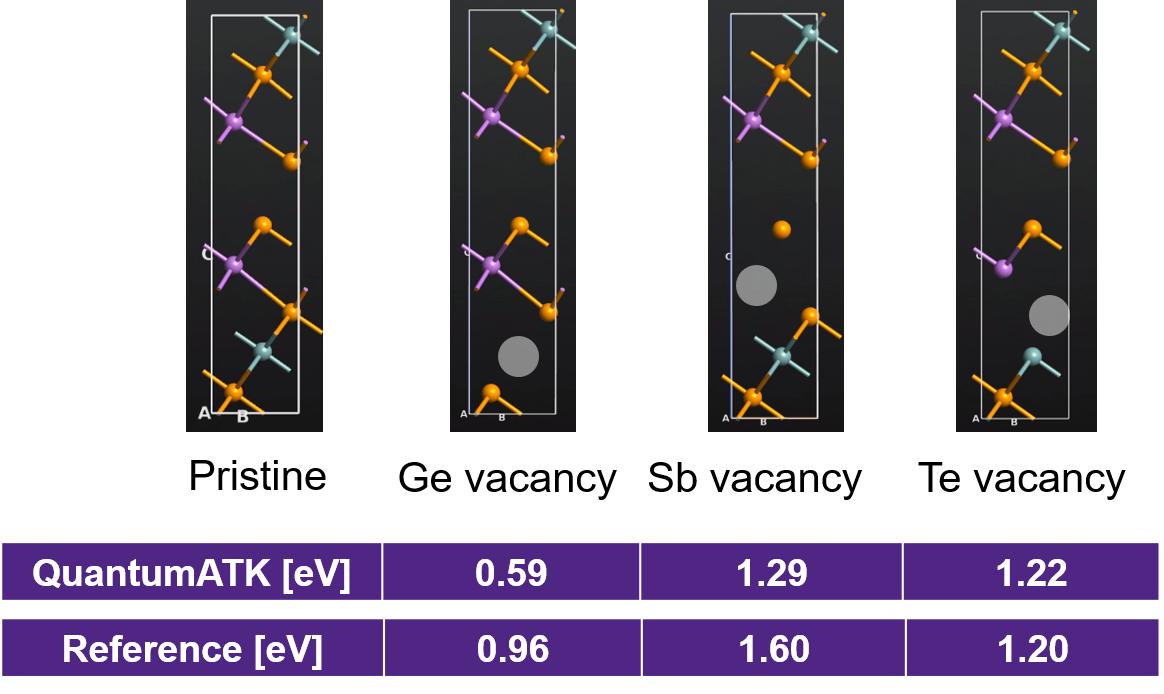
We perform three vacancy calculations by removing either one Ge, one Sb or one Te atom from the unitcell geometry as highlighted in the above image and replacing them with a ghost atom.
A ghost atom is used to include the basis functions for that removed atom in the calculation.
The corresponding scripts are given here (Ge-vacancy, Sb-vacancy, Te-vacancy).
As can be seen from the above image, the formation energy of Ge vacancy is the lowest followed by Te and Sb.
This ordering is similar to the literature results with some discrepancies in the absolute values.
This could be attributed to the different lattice parameters and basis sets (reference: plane wave basis and PAW, QuantumATK: LCAO and PseudoDojo medium) used in the literature reference and in our calculations.
Total DOS With and Without Ge Vacancy¶
In this section we compare the total density of states from the GST (5x1x1) supercell with and without a Ge vacancy and compare the results to literature [4].
The scripts used for this calculation can be downloaded from these links (Pristine, VGe).
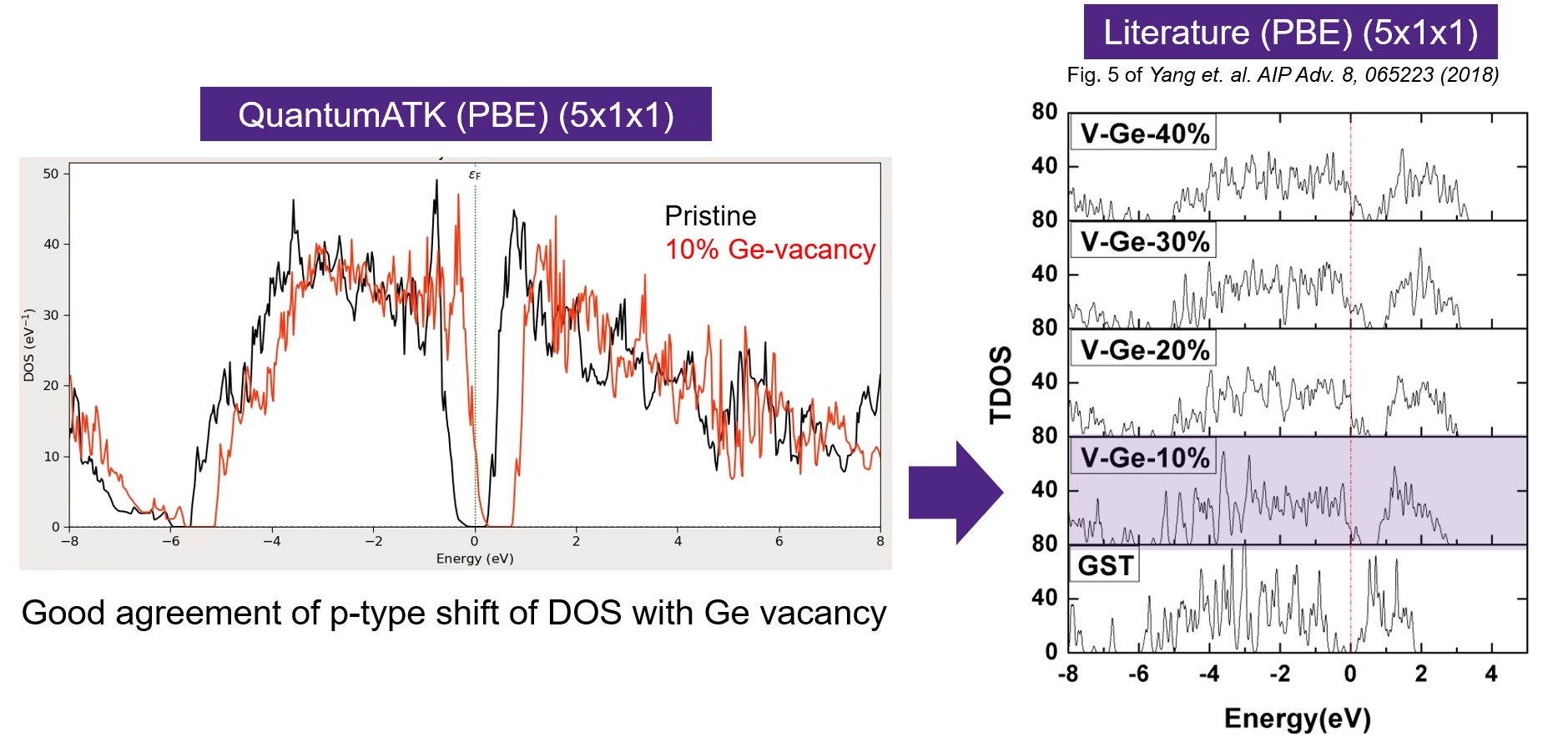
The above plot shows the DOS comparison and a clear p-type shift is observed on including 10% Ge vacancy. This observation is in good qualitative agreement with the reference.