DynamicalMatrix¶
Included in QATK.Analysis
- class DynamicalMatrix(configuration, filename, object_id, calculator=None, repetitions=None, atomic_displacement=None, acoustic_sum_rule=None, finite_difference_method=None, constraints=None, constrain_electrodes=None, use_equivalent_bulk=None, max_interaction_range=None, force_tolerance=None, processes_per_displacement=None, log_filename_prefix=None, use_wigner_seitz_scheme=None, use_symmetry=None, polar_phonon_splitting_parameters=None, use_internal_coordinates=None, bond_list=None)¶
Constructor for the DynamicalMatrix object.
- Parameters:
configuration (
BulkConfiguration|MoleculeConfiguration|DeviceConfiguration) – The configuration for which to calculate the dynamical matrix.calculator (
Calculators) – The calculator to be used in the dynamical matrix calculations. Default: The calculator attached to the configuration.filename (str) – The full or relative path to save the results to. See
nlsave().object_id (str) – The object id to use when saving. See
nlsave().repetitions (
Automatic| list of ints) – The number of repetitions of the system in the A, B, and C-directions given as a list of three positive integers, e.g.[3, 3, 3], orAutomatic. Each repetition value must be odd. Default:Automaticatomic_displacement (PhysicalQuantity of type length) – The distance the atoms are displaced in the finite difference method. Default:
0.01 * Angstromacoustic_sum_rule (bool) – Control if the acoustic sum rule should be invoked. Default:
Truefinite_difference_method (
Forward|Central) – The finite difference scheme to use. Default:Centralconstraints (list of type int) – List of atomic indices that will be constrained, e.g.
[0, 2, 10]. Default: Noneconstrain_electrodes (bool) – Control if the electrodes and electrode extensions should be constrained in case of a
DeviceConfiguration. Default:Falseuse_equivalent_bulk (bool) – Control if a
DeviceConfigurationshould be treated as aBulkConfiguration. Default:Truemax_interaction_range (PhysicalQuantity of type length) – Set the maximum range of the interactions. Default: All atoms are included
force_tolerance (PhysicalQuantity of type energy per length squared) – All force constants below this value will be truncated to zero. Default:
1e-8 * Hartree/Bohr**2processes_per_displacement (int |
AllProcesses|ProcessesPerNode) – The number of processes assigned to calculating a single displacement. Default:1for force-field calculators,AllProcessesotherwise.log_filename_prefix (str or None) – Prefix for the filenames where the logging output for every displacement calculation is stored. The filenames are formed by appending a number and the file extension (“.log”). If a value of None is given then all logging output is done to stdout. If a classical calculator is used, no per-displacment log files will be generated. Default:
"forces_displacement_"use_wigner_seitz_scheme (bool) – Control if the real space Dynamical Matrix should be extended according to the Wigner Seitz construction.
use_wigner_seitz_scheme=True. Default:Falsefor ForceField calculator,Trueotherwise.use_symmetry (bool) – If enabled, only the symmetrically unique atoms are displaced and the remaining force constants are calculated using symmetry. Default:
Truepolar_phonon_splitting_parameters (
PolarPhononSplittingParameters|None) – If given polar phonon splitting will be included in the dynamical matrix. An optical_spectrum and born_effective_charge tasks will be added to the work flow. Default:None, i.e. polar phonon splitting is not included.use_internal_coordinates (bool) – Control if the eigensystem should be solved in internal coordinates for a
MoleculeConfigurationDefault:Falsebond_list (list of list of type int) – List of og sublists with bonding atomic indices that will be used to generate internal coordinates, e.g.
[[0, 1], [0, 2]]. Default:None
- acousticSumRule()¶
- Returns:
Return if the acoustic sum rule is invoked.
- Return type:
bool
- atomicDisplacement()¶
- Returns:
The distance the atoms are displaced in the finite difference method.
- Return type:
PhysicalQuantity with length unit
- bondList()¶
- Returns:
The bond_list optionally supplied for an internal coordinate scheme calculation.
- Return type:
list | None
- constrainElectrodes()¶
- Returns:
Boolean determining if the electrodes and electrode extensions are constrained in case of a DeviceConfiguration.
- Return type:
bool
- constraints()¶
- Returns:
The list of constrained atoms.
- Return type:
list of int
- dependentStudies()¶
- Returns:
The list of dependent studies.
- Return type:
list of
Study
- enablePolarPhononSplitting()¶
- Returns:
Return true if the polar phonon splitting is enabled.
- Return type:
bool
- filename()¶
- Returns:
The filename where the study object is stored.
- Return type:
str
- finiteDifferenceMethod()¶
- Returns:
The finite difference scheme to use.
- Return type:
Central|Forward
- forceTolerance()¶
- Returns:
The force tolerance
- Return type:
PhysicalQuantity with an energy per length squared unit e.g. Hartree/Bohr**2
- logFilenamePrefix()¶
- Returns:
The filename prefix for the logging output of the study.
- Return type:
str |
LogToStdOut
- maxInteractionRange()¶
- Returns:
The maximum interaction range.
- Return type:
PhysicalQuantity with length unit
- nlinfo()¶
- Returns:
Structured information about the DynamicalMatrix.
- Return type:
dict
- nlprint(stream=None)¶
Print a string containing an ASCII table useful for plotting the Study object.
- Parameters:
stream (python stream) – The stream the table should be written to. Default:
NLPrintLogger()
- numberOfProcessesPerTask()¶
- Returns:
The number of processes to be used to execute each task. If None, all available processes execute each task collaboratively.
- Return type:
int | None |
ProcessesPerNode
- numberOfProcessesPerTaskResolved()¶
- Returns:
The number of processes to be used to execute each task. Default values are resolved based on the current execution settings.
- Return type:
int
- objectId()¶
- Returns:
The name of the study object in the file.
- Return type:
str
- phononEigensystem(q_point=None, constrained_atoms=None, enable_polar_phonon_splitting=None, D=None)¶
Calculate the eigenvalues and eigenvectors for the dynamical matrix at a specified q-point.
- Parameters:
q_point (list of 3 floats) – The fractional q-point to use. Default:
[0.0, 0.0, 0.0]constrained_atoms (list of ints.) – List of atoms being constrained. The matrix elements from these atoms will not be included in the calculation of the eigensystem. Default: [] (empty list)
enable_polar_phonon_splitting (bool) – Boolean controling if polar optical splitting should be included.
- Returns:
The eigenvalues and eigenvectors of the dynamical matrix.
- Return type:
2-tuple containing the eigenvalues and eigenvectors
- polarPhononSplittingParameters()¶
- Returns:
The polar optical splitting parameters.
- Return type:
PolarOpticalSplittingParameters
- processesPerDisplacement()¶
- Returns:
The number of processes per displacement.
- Return type:
int |
AllProcesses|ProcessesPerNode
- realSpaceDynamicalMatrix()¶
Returns the real space dynamical matrix. The shape of the matrix is (N, M), where N is the number of degrees of freedom (3 * number of atoms), and M = N * R, where R is the total number of repetitions.
Each subblock D[i*N:(i+1)*N, :] corresponds to the matrix elements between the center block (where the atoms have been displaced) and a neighbouring cell translated from the central cell by translations[i] in fractional coordinates.
- Returns:
The real-space dynamical matrix as a sparse matrix together with a list of translation vectors in fractional coordinates. The real space dynamical matrix is given in units of (meV / hbar)**2.
- Return type:
(scipy.sparse.csr_matrix, list of list(3) of integers)
- reciprocalSpaceDynamicalMatrix(q_point=None)¶
Evaluate the reciprocal space dynamical matrix for a given q-point in reciprocal space.
- Parameters:
q_point (list of floats) – The fractional q-point to use. Default:
[0.0, 0.0, 0.0]- Returns:
The dynamical matrix for
q_point.- Return type:
PhysicalQuantity of units
(meV / hbar)**2
- repetitions()¶
- Returns:
The number of repetitions in the A, B, and C-directions for the supercell that is used in the finite displacement calculation.
- Return type:
list of three int.
- saveToFileAfterUpdate()¶
- Returns:
Whether the study is automatically saved after it is updated.
- Return type:
bool
- symmetry()¶
- Returns:
True if the use of crystal symmetry to reduce the number of displacements is enabled.
- Return type:
bool
- uniqueString()¶
Return a unique string representing the state of the object.
- update()¶
Run the calculations for the DynamicalMatrix study object.
- useEquivalentBulk()¶
- Returns:
Boolean determining if a DeviceConfiguration is treated as a BulkConfiguration.
- Return type:
bool
- useInternalCoordinates()¶
- Returns:
Return True if the use of internal coordinates is enabled.
- Return type:
bool
- wignerSeitzScheme()¶
- Returns:
Boolean to control if the real space Dynamical Matrix should be extended according to the Wigner-Seitz construction.
- Return type:
bool
Usage Examples¶
Note
Study objects behave differently from analysis objects. See the Study object overview for more details.
Calculate the DynamicalMatrix for a system repeated five times in the B direction and three times in the C direction.
dynamical_matrix = DynamicalMatrix(
configuration,
filename='DynamicalMatrix.hdf5',
object_id='dynamical_matrix',
repetitions=(1,5,3)
)
dynamical_matrix.update()
When using repetitions=Automatic, the cell is repeated such that all atoms within a pre-defined, element-pair dependent interaction range are included.
dynamical_matrix = DynamicalMatrix(
configuration,
filename='DynamicalMatrix.hdf5',
object_id='dynamical_matrix',
repetitions=Automatic
)
dynamical_matrix.update()
The default number of repetitions i.e. repetitions=Automatic can be found before a calculation using the function
checkNumberOfRepetitions().
(nA, nB, nC) = checkNumberOfRepetitions(configuration)
The maximum interaction range between two atoms can be specified manually, by using
the max_interaction_range keyword.
dynamical_matrix = DynamicalMatrix(
configuration,
filename='DynamicalMatrix.hdf5',
object_id='dynamical_matrix',
repetitions=Automatic,
max_interaction_range=12.0*Ang,
)
dynamical_matrix.update()
Notes¶
The DynamicalMatrix is calculated using the finite difference
method in a repeated cell, which is sometimes also referred to as frozen-
phonon or super-cell method.
In the following, we denote the atoms in the unit cell by \(\mu\) and the atoms in the repeated cell by \(i\). Furthermore, denote the dynamical matrix elements, \(D_{\mu \alpha, i \beta}\), where \(\alpha, \beta\) are the Cartesian directions, i.e. \(x, y, z\). A dynamical matrix element is given by
where \(F_{i \beta}\) is the force on atom \(i\) in direction \(\beta\) due to a displacement of atom \(\mu\) in direction \(\alpha\).
The derivative is calculated by either forward or central finite
differences, where in the following we will focus on the latter. Atom
\(\mu\) is displaced by \(\Delta r_\alpha\) and \(-\Delta
r_\alpha\), and the changes in the force, \(\Delta F_{i \beta}\) are
calculated to approximate the dynamical matrix element
The default is to use repetitions=Automatic. In this case the cell is
repeated such that all atoms within a pre-defined element-dependent distance
from the atoms in the unit cell are included in the repeated cell. The
repetitions used is written to the output file. These default interaction
ranges are suitable for most systems.
However, if you are using long-ranged interactions, e.g. classical potentials
with electrostatic interactions in TremoloXCalculator, it might be
necessary to increase the number of repetitions.
For a 1D or 2D system, the unit cell should not be repeated in the confined
directions. This is only discovered by the repetitions=Automatic, if there
is enough vacuum in the unit cell in the directions that should not be
repeated. That is typically 10-20 Å vacuum depending on the elements and their
interaction ranges. Thus, for confined systems it is recommended to check the
repetitions used and possibly use manual instead of automatic repetition.
DynamicalMatrix calculations using DFT or Semi-Empirical calculators have
functionality to fully resume partially completed calculations by re-running the
same script or reading the study object from file and calling update() on
it. The study object will automatically detect which displacement
calculations have already been carried out and only run the ones that are not
yet completed. To ensure highest performance this functionality is not available
for ATK-ForceField calculations.
Notes for DFT¶
In ATK-DFT the number of repetitions of the unit cell in super cell must ensure
that the change in the force on atoms outside the super cell is zero for every
atomic displacement in the center cell. An equivalent discussion of the number
of k-points of the super cell and the number of repetitions can be found for
HamiltonianDerivatives in the section Notes for DFT.
Consider a system with e.g. \(x\) and \(y\) as confined directions and
the k-point sampling of the unit cell \((1, 1, N_{\text{C}})\), see
Fig. 179 (a). Assume that the number of
repetitions in the C-direction is known for the change in the force on atoms
outside the super cell to be zero. Then the number of repetitions must be
\((1, 1, {\scriptstyle \text{repetitions in C}})\). Furthermore, the k-point
sampling of the super cell becomes \((1, 1, \frac{N_{\text{C}}}{\text{repetitions in C}})\).
Note
From QuantumATK-2019.03 onwards, the k-point sampling and
density-mesh-cutoff will be automatically adapted to the given number
of repetitions when setting up the super cell inside
DynamicalMatrix and HamiltonianDerivatives. That
means you can specify the calculator settings for the unit cell and use it
with any desired number of repetitions in dynamical matrix and hamiltonian
derivatives calculations.
When calculating the DynamicalMatrix with ATK-DFT, accurate results
may require a higher precision than usual by increasing the density_mesh_cutoff
in NumericalAccuracyParameters and decreasing the tolerance in
IterationControlParameters, e.g.
numerical_accuracy_parameters = NumericalAccuracyParameters(
density_mesh_cutoff=150.0*Hartree
)
iteration_control_parameters = IterationControlParameters(
tolerance=1e-6
)
Notes for the simplified supercell method (use_wigner_seitz_scheme=True)¶
The simplified supercell method is an approximation which allows to obtain the
dispersion of vibrational eigenmodes with a force calculation of the unit cell
only, i.e. having repetitions=[1,1,1] (the poor man’s frozen phonon
calculation). It is valid if the unit cell is large enough, i.e. if it
accommodates 200 atoms and more. The convergence should be checked with
respect to the number of atoms per unit cell or by a conventional frozen
phonon calculation.
Idea of the simplified supercell method¶
In large unit cells, the force of atom i due to a displacement of atom j is small for distances of half the length of the unit cell vectors. Due to the translational invariance, a displaced atom i contributes from two sides to the force on atom j, which can be exactly decomposed if the distance between the atoms is large enough. As an example, we will look at a 1D chain with 6 atoms per unit cell (see Fig. 172).
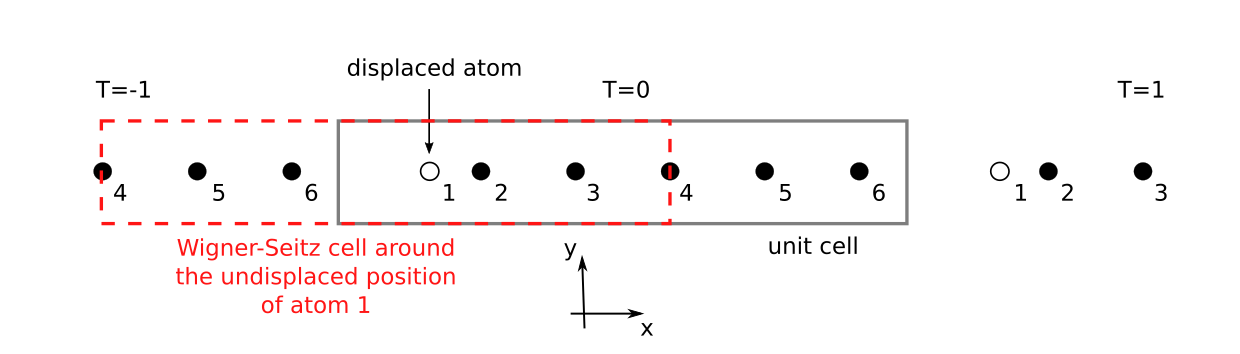
Fig. 172 A 1D chain with 6 atoms per unit cell with atom 1 being displaced. The periodically-repeated atoms to the left (\(T=-1\)) and to the right (\(T=1\)) are indicated. The Wigner-Seitz cell centered at the undisplaced position of atom 1 is drawn red / dashed.¶
All atoms are at their equilibrium positions, besides atom 1 which is
displaced along the \(x\) direction. The force on atom 5, for example,
can be regarded as “repulsive” due to the displacement of atom 1 in the unit
cell with translation \(T=0\) along the \(x\)-axis. However, there is
also an “attractive” contribution from the image of the displaced atom 1 in
the unit cell with translation \(T=1\). To decompose the two contributions
we construct a Wigner-Seitz cell centered at the undisplaced position of atom
1 and check the distance to the nearest neighbor representations of atom 5.
The representation of atom 5 inside the cell at \(T=0\) is not part of the
Wigner-Seitz cell, and thus this contribution is neglected. In contrast, the
representation of atom 5 at \(T=-1\) is inside the Wigner-Seitz cell and
we keep this contribution. If an atom is at the face (like atom 4), the edge
or a vertex of the Wigner-Seitz cell, the force is included weighted by the
inverse multiplicity of this Wigner-Seitz grid point. This construction is
repeated for all atoms inside the unit cell. In this way it is possible to
approximately calculate the entries of the DynamicalMatrix for all
repetitions of the cell from \(T = -1\) to \(T = 1\) and thereby get
the dispersion, by only calculating the forces in a single unit cell.
Note
The Wigner-Seitz construction ensures that the phonon spectrum at the \(\Gamma\)-point is preserved. Hence, a separate \(\Gamma\)-point calculation is not necessary.
Notes for polar phonon splitting¶
In polar materials the covalency will change during ionic displacements since a macroscopic electric field is generated. This gives rise to an extra force constant term in the dynamical matrix. The full dynamical matrix is \(\mathbf{D}(\mathbf{q}) = \mathbf{D}^{FC}(\mathbf{q}) + \mathbf{D}^{NA}(\mathbf{q})\). Here \(\mathbf{D}^{FC}(\mathbf{q})\) is the usual force constant dynamical matrix discussed above and the second term \(\mathbf{D}^{NA}(\mathbf{q})\) is due to polar phonon interaction. The non-analytic (NA) contribution from the macroscopic field to the dynamical matrix is:
Here \(\mathbf{Z}(i)\) is the Born effective charge tensor (a matrix for atom \(i\)), \(\hat{\mathbf{q}}=\mathbf{q}/|\mathbf{q}|\) is a unit vector, \(M_i\) is the mass of atom \(i\), \(\Omega\) is the unit cell volume, \(e\) is the electron charge and \(\epsilon_{0}\) is the vacuum permittivity.
The term non-analytical refers to the fact that it approaches different values from different \(\mathbf{q}\)-space directions as \(q\rightarrow 0\).
It is obtained from the BornEffectiveCharge calculation and the high-frequency dielectric tensor \(\epsilon^{\infty}\) obtained from the OpticalSpectrum.
The Dynamical Matrix study object will automatically include a calculation of the BornEffectiveCharges and OpticalSpectrum or it can be given by the user from an existing calculation.
To include polar phonon splitting in the dynamical matrix study object one has to give PolarPhonon_splitting_parameters as input:
dynamical_matrix = DynamicalMatrix(
configuration,
filename='DynamicalMatrix.hdf5',
object_id='dynamical_matrix',
repetitions=(1,5,3),
polar_phonon_splitting_parameters=pps_parameters,
)
dynamical_matrix.update()
Here the polar polar_phonon_splitting_parameters is a container of polar phonon splitting parameters. It can either be setup by defaults
pps_parameters = PolarPhononSplittingParameters()
Or it can be set with specific user-defined variables for optical spectrum and Born effective charges.
pps_parameters = PolarPhononSplittingParameters(
kpoints=MonkhorstPackGrid(25, 25, 25),
broadening=0.1 * eV,
bands_below_fermi_level=35,
bands_above_fermi_level=35,
kpoints_a=MonkhorstPackGrid(19, 9, 9),
kpoints_b=MonkhorstPackGrid(9, 19, 9),
kpoints_c=MonkhorstPackGrid(9, 9, 19),
atomic_displacement=0.01 * Ang,
)
Finally it can be defined from existing calculations of the OpticalSpectrum and BornEffectiveCharge:
pps_parameters = PolarPhononSplittingParameters(
optical_spectrum=optical_spectrum,
born_effective_charge=born_effective_charge,
)
This last option enables the combination of force constants obtained from a classical calculator with polar phonon splitting obtained from ATK-DFT.
Notes for internal coordinates¶
For molecules, it can be beneficial to shift the dynamical matrix into an internal coordinate
basis when conducting studies of or including the vibrational modes of a
MoleculeConfiguration. A (potentially) redundant internal coordinate basis is generated
by converting the bond connectivity of the molecule into unique stretch, bend, and dihedral
internal coordinates, from which a non-redundant set of \(3N-6\) (in the general case) linear
combinations of internal coordinates are derived. This scheme thus allows a non-redundant description
of the vibrational modes and results in the same number of vibrational modes as the number of degrees
of freedom in the molecule.
The internal coordinate scheme assumes that the bond connectivity of the system connects all atoms
in the configuration, so that at least \(3N-6\) unique internal coordinates are generated. If
fewer bonds are present, or if only a smaller number of bonds can be automatically discovered, the
internal coordinate scheme will yield fewer vibrational modes than the degrees of freedom in the
molecule. The use_internal_coordinates argument should therefore only be activated when all
bonds in a molecule can be automatically discovered with the findBonds() method of a
MoleculeConfiguration object or are set manually on the configuration. This can be done
from the Builder or in scripting. Alternatively, a bond_list argument can be passed to a
DynamicalMatrix object with the desired bonding behaviour, in which all atom indices are
included and interconnected in a single molecule fragment.