AdsorbedSurfaceGenerator¶
Included in QATK.Surfaces
- class AdsorbedSurfaceGenerator(substrate=None, search_depth=None, surface_level=None, grouping_precision=None)¶
A data structure for keeping track of the input needed to create an adsorbed surface.
- Parameters:
substrate (
BulkConfiguration|SurfaceConfiguration) – Optionally provide a substrate.search_depth (PhysicalQuantity of type length) – The depth below the surface (defined from the highest atom in the configuration) at which atoms are included in defining adsorption sites.
surface_level (PhysicalQuantity of type length) – The atomic plane in the substrate below which to consider surface.
grouping_precision (float) – The precision of grouping unique adsorption sites. Higher values result in more adsorption sites. The allowed range is [0, 2].
- addAnchorAtom(molecule_key, atom_index, site_type=None, site_index=<class 'NL.ComputerScienceUtilities.NLFlag._NLFlag.Random'>)¶
Add an anchor atom to the molecule type referenced by molecule_key.
- Parameters:
molecule_key (str) – A unique key specifying a molecule type.
atom_index (int) – The atom index of the anchor atom.
site_type (str) – The site type that the anchor atom should occupy.
site_index (Random | int) – A site_index from the given site type that this anchor atom should occupy.
- addMolecule(molecule_key, molecule, site_type=None, distribution=<class 'NL.ComputerScienceUtilities.NLFlag._NLFlag.Random'>, count=0, occupancy=0.0, height=PhysicalQuantity(2.0, Ang), orientation=0, anchor_atom_index=0, gridcell_repetitions=(1, 1), gridcell_center=False)¶
Add a molecule type to the surface.
- Parameters:
molecule_key (str) – A unique key specifying a molecule type.
molecule (
MoleculeConfiguration) – A molecule to be added to the surface.site_type (str) – The site type that the molecule’s main anchor atom should occupy.
distribution (int | Random | Quasirandom | SurfaceGrid) – The distribution for the molecule, which is also the distribution of the main anchor atom. Should be a site index for the given site_type or a flag.
count (int) – The number of molecules of this type.
occupancy (float) – The occupancy of this molecule type on the main_anchor_atom_site_type. The allowed range is from 0.0 (a clean surface) to 1.0 (maximum site occupation).
height (PhysicalQuantity of type length) – The height of the molecules of this type.
orientation (ORIENTATION | ORIENTATIONS) – The orientation for this molecule type.
anchor_atom_index (int) – The atom index for the main anchor atom.
gridcell_repetitions (tuple of two integers) – If distributing to SurfaceGrid, the repetitions of the grid.
gridcell_center (bool) – If distributing to SurfaceGrid, whether to use the center of grid cells.
- adsorptionSiteParameters()¶
- Returns:
The parameters used to find the current adsorption sites as a tuple of the search_depth, surface_level and grouping_precision, in that order.
- Return type:
tuple
- adsorptionSiteTypes()¶
- Returns:
The names of all available adsorption site types.
- Return type:
tuple
- adsorptionSites()¶
- Returns:
The adsorption sites as a dict, where the keys are site types and the values are lists of coordinates for the adsorption sites.
- Return type:
dict
- changeAnchorAtom(molecule_key, index, atom_index=None, site_type=None, site_index=None)¶
Change an anchor atom in the molecule type referenced by molecule_key.
- Parameters:
molecule_key (str) – A unique key specifying a molecule type.
index (int) – The index of the anchor atom to change.
atom_index (int) – The atom index of the anchor atom.
site_type (str) – The site type that the anchor atom should occupy.
site_index (Random | SurfaceGrid | Quasirandom | int) – A distribution flag or a site_index from the given site type that this anchor atom should occupy.
- changeMolecule(molecule_key, new_molecule_key=None, molecule=None, site_type=None, distribution=None, count=None, occupancy=None, height=None, orientation=None, anchor_atom_index=None, gridcell_repetitions=None, gridcell_center=None)¶
Change a molecule type.
- Parameters:
molecule_key (str) – A unique key specifying a molecule type.
new_molecule_key (str) – A new molecule key.
molecule (
MoleculeConfiguration) – The molecule being changed.site_type (str) – The site type that the molecule’s main anchor atom should occupy.
distribution (int | Random | Quasirandom | SurfaceGrid) – The distribution for the molecule, which is also the distribution of the main anchor atom. This should be a site index for the given site_type or a flag.
count (int) – The number of molecules of this type.
occupancy (float) – The occupancy of this molecule type on the main_anchor_atom_site_type. Range: [0.0, 1.0].
height (PhysicalQuantity of type length) – The height of the molecules of this type.
orientation (ORIENTATION | ORIENTATIONS) – The orientation for this molecule type.
anchor_atom_index (int) – The atom index for the main anchor atom.
gridcell_repetitions (tuple of two integers) – If distributing to SurfaceGrid, the repetitions of the grid.
gridcell_center (bool) – If distributing to SurfaceGrid, whether to use the center of grid cells.
- copyMoleculeTypes(other, molecule_key=None)¶
Copy molecule types from another instance of AdsorbedSurfaceGenerator into this instance. If molecule_key is given, a copy of a single molecule type corresponding to the molecule_key is made, else all molecule types are copied. Note that this does not copy the site_type and distribution for any anchor atoms, as they may not match the adsorption sites in this instance, and should be set explicitly after copying.
- Parameters:
other (AdsorbedSurfaceGenerator) – The AdsorbedSurfaceGenerator to copy from.
molecule_key (str) – A specific molecule_key.
- findAdsorptionSites(search_depth=PhysicalQuantity(0.9, Ang), surface_level=None, grouping_precision=1.0)¶
Find adsorption sites by defining a volume with the dimensions cell vector A, cell vector B and ‘search_depth’. All atoms within this volume are assumed to constitute the substrate surface and will be used to generate adsorption sites.
- Parameters:
search_depth (PhysicalQuantity of type length) – The depth below the surface (defined from the highest atom in the configuration) at which atoms are included in defining adsorption sites.
surface_level (PhysicalQuantity of type length | None) – The atomic plane in the substrate below which to consider surface. Default: The highest atom is considered the surface level.
grouping_precision (float) – The precision of grouping unique adsorption sites. Higher values result in more adsorption sites. The allowed range is [0, 2].
- Returns:
The adsorption sites.
- Return type:
dict
- generateAdsorbedSurface(fit_cell=None, bottom_vacuum=None, top_vacuum=None, optimize_structure=None, random_seed=None)¶
Create a configuration consisting of a substrate with adsorbed molecules.
- Parameters:
fit_cell (bool) – Fit the cell in the transverse plane to ensure that the molecules does not wrap around the boundaries and bond to itself and/or other molecules. Default: False
bottom_vacuum (PhysicalQuantity of type length) – The bottom vacuum height in the projected C-direction. Default: The original bottom vacuum of the configuration.
top_vacuum (PhysicalQuantity of type length) – The top vacuum height in the projected C-direction. Default: The original top vacuum of the configuration.
optimize_structure (bool) – Whether to refine the molecules on the surface. Default: False
random_seed (int | None) – Optionally, provide a random seed for generating random molecule placements.
- Returns:
A configuration consisting of the substrate with adsorbed molecules.
- Return type:
- moleculeKeys()¶
- Returns:
The keys of all molecule types added.
- Return type:
tuple
- numberOfAnchorAtoms(molecule_key)¶
Get the number of anchor atoms on the molecule type referenced by molecule_key.
- Parameters:
molecule_key (str) – The key of the molecule of interest.
- Returns:
The number of anchor atoms.
- Return type:
int
- static overlappingMolecules(configuration, tolerance=PhysicalQuantity(1.0, Ang))¶
Find overlapping molecules. The premise of this method is as follows:
Find all pairs of overlapping atoms. These pairs are unique and do not contain mirrored copies, i.e., only one of [0, 1] and [1, 0] will be in the list.
Collect molecule tags for relevant overlaps by extracting the tags for each pair of overlapping atoms.
Ignore any that involve the substrate.
Ignore tags that are were not added by AdsorbedSurfaceGenerator by using the prefix ‘adsmol’.
Intramolecular ‘overlaps’ are false positives, so we ignore these as well. These will have the same molecular tag on both atoms.
By default, only take the second molecule tag in a pair of overlapping molecules, since only one of them needs to be moved to fix the overlap. If take_both=True, then both tags are taken (This is useful if we need to check if a particular molecule is overlapping any other).
- Parameters:
configuration (BulkConfiguration) – The adsorbed surface configuration.
tolerance (
PhysicalQuantityof type distance) – The intermolecule distance tolerance. Default: 1 * Angstrom.
- Returns:
The set of unique tags for overlapping molecules.
- Return type:
list
- raiseDistributionError(message)¶
Reset the site distribution and raise NLValueError.
- Parameters:
message (str) – The error message.
- redistributeOverlappingMolecules(configuration, tolerance=PhysicalQuantity(1.0, Ang))¶
Try to redistribute overlapping molecules to remove the overlaps.
Note! This only applies to molecules distributed with the Random distribution.
The premise of this method is as follows:
1. Find molecules that need to be moved due to atomic overlaps with other molecules. The substrate is ignored since there are better ways to avoid molecules cutting into the surface.
2. If any, then loop over the found molecules and for each of them: a. Loop over all free sites for the molecule’s site type b. Place the molecule and check if it overlaps anything. c. If yes, then check the next free site. d. If not, then accept the position and move on to the next molecule.
3. If all free sites were checked and none of them satisfied the distance criteria, the molecule remains at its original adsorption site. —.
- Parameters:
configuration (BulkConfiguration) – The adsorbed surface configuration.
tolerance (
PhysicalQuantityof type distance) – The inter-molecule distance tolerance. Default: 1 * Angstrom.
- Returns:
The adjusted configuration.
- Return type:
- removeAnchorAtom(molecule_key, index)¶
Remove an anchor atom in the molecule given by molecule_key.
- Parameters:
molecule_key (str) – A unique key specifying a molecule type.
index (int) – The index of the anchor atom to remove.
- removeMolecule(molecule_key, redistribute=False)¶
Remove a molecule type.
- Parameters:
molecule_key (str) – A unique key specifying a molecule type.
- setSubstrate(substrate, bottom_vacuum=None, top_vacuum=None)¶
Set a substrate if the current substrate is None. SurfaceConfigurations will be converted into BulkConfiguration.
- Parameters:
substrate (
BulkConfiguration|SurfaceConfiguration) – The substrate.bottom_vacuum (PhysicalQuantity of type length) – The bottom vacuum height in the projected C-direction. Default: The original bottom vacuum of the configuration.
top_vacuum (PhysicalQuantity of type length) – The top vacuum height in the projected C-direction. Default: The original top vacuum of the configuration.
- substrate()¶
- Returns:
The configuration used as the substrate, which is a surface slab with vacuum.
- Return type:
- uniqueString()¶
Return a unique string representing the state of the object.
Usage Examples¶
Example 1. Adsorb water molecules onto the ontop sites of a Pt(100) surface at 50% coverage, anchored by their oxygen atoms and oriented vertically.
from example_configurations import substrate, water
# -------------------------------------------------------------
# Create surface with adsorbed molecules.
# -------------------------------------------------------------
# Instantiate the AdsorbedSurfaceGenerator with the substrate and find adsorption sites.
generator = AdsorbedSurfaceGenerator(
substrate=substrate,
search_depth=0.9 * Angstrom,
distance=1.0 * Angstrom,
grouping_precision=1.0
)
# Add molecules.
generator.addMolecule(
molecule_key='Water',
molecule=water,
count=9,
orientation=ORIENTATIONS.VERTICAL,
anchor_atom_index=1,
site_type='ontop_Platinum',
distribution=Random
)
# Generate a configuration with the molecules adsorbed to the substrate surface.
configuration = generator.generateAdsorbedSurface()
adsorbed_surface_example1.py
example_configurations.py
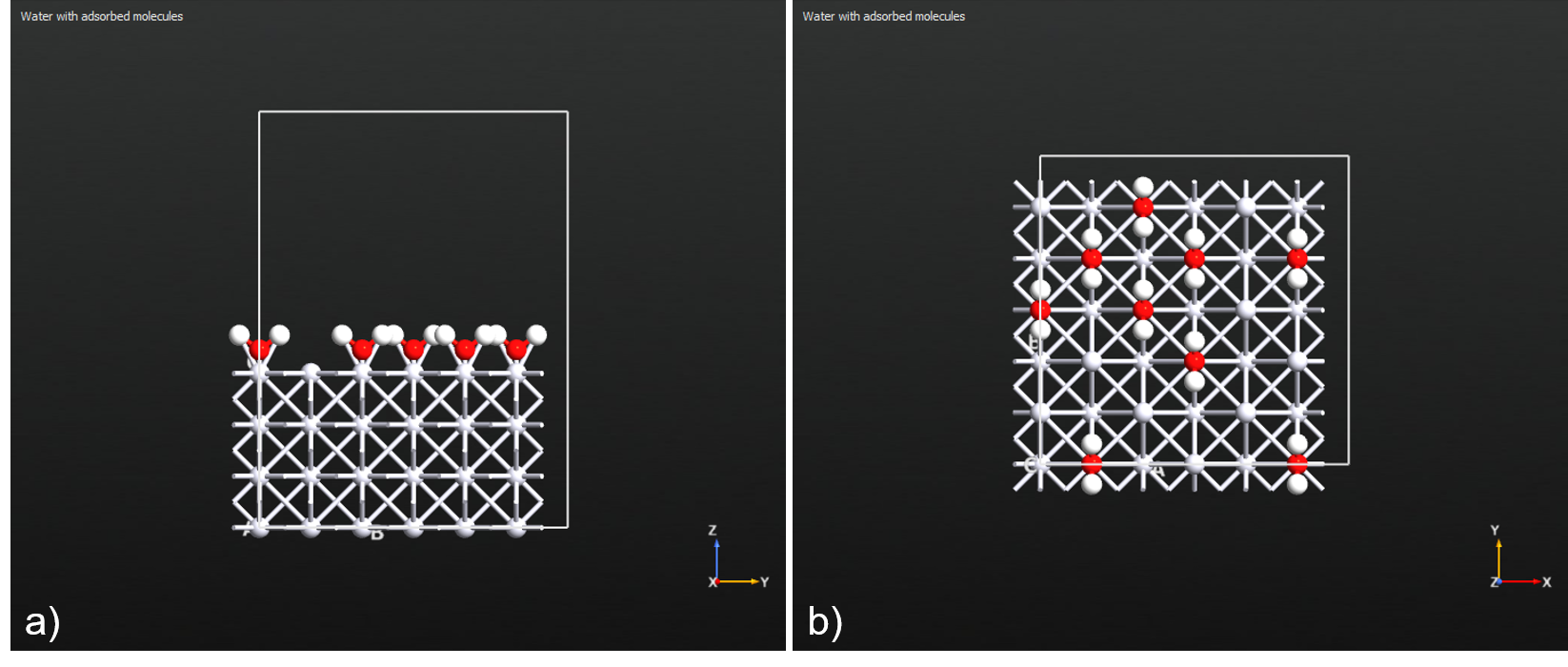
Fig. 161 Resulting configuration from Example 1. a) Side view. b) Top view.¶
Example 2. Adsorb water molecules onto the ontop sites of a Pt(100) surface at 50% coverage, anchored by both of their hydrogen atoms and oriented with the second anchor atom on bridge sites.
from example_configurations import substrate, water
# -------------------------------------------------------------
# Create surface with adsorbed molecules.
# -------------------------------------------------------------
# Instantiate the AdsorbedSurfaceGenerator with the substrate and find adsorption sites.
generator = AdsorbedSurfaceGenerator(
substrate=substrate,
search_depth=0.9 * Angstrom,
distance=1.0 * Angstrom,
grouping_precision=1.0
)
# Add molecules.
generator.addMolecule(
molecule_key='Water',
molecule=water,
count=9,
orientation=ORIENTATIONS.ANCHORED,
anchor_atom_index=0,
site_type='ontop_Platinum',
distribution=Random
)
generator.addAnchorAtom(
molecule_key='Water',
atom_index=2,
site_type='bridge',
distribution=Random
)
# Generate a configuration with the molecules adsorbed to the substrate surface.
configuration = generator.generateAdsorbedSurface()
adsorbed_surface_example2.py
example_configurations.py
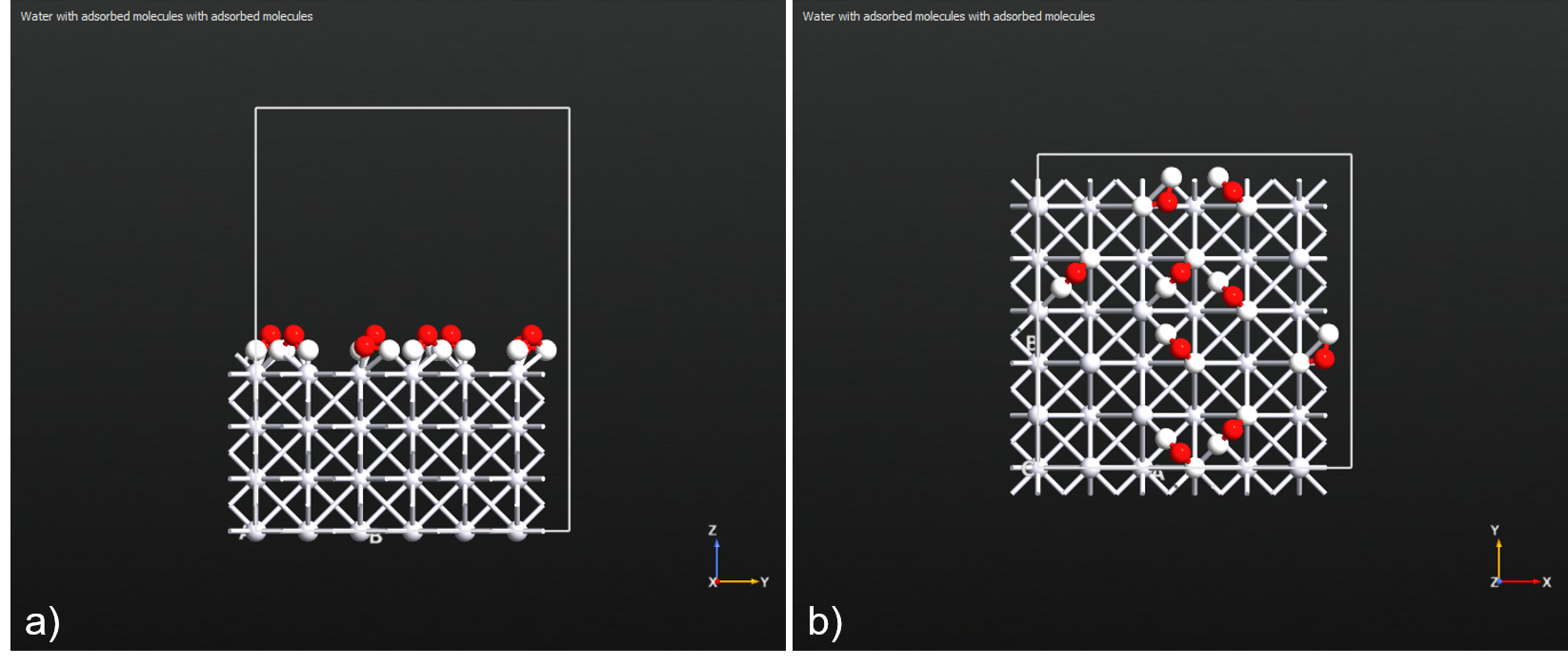
Fig. 162 Resulting configuration from Example 2. a) Side view. b) Top view.¶
General¶
The AdsorbedSurfaceGenerator class is designed to collect the necessary components for creating a surface with adsorbed molecules, as well as to modify these components and generate the final configuration. The basic steps of using the class are:
Add a substrate.
Calculate adsorption sites for the substrate.
Add molecule types and define how they should be distributed onto the substrate.
Optionally add additional anchor atoms.
Call
generateAdsorbedSurfaceto create the final configuration.
The class can be instantiated with a substrate configuration and parameters for finding adsorption sites as optional input arguments. This allows for a quick start-up by setting the substrate and calculating the adsorption sites in one go.
Substrate and adsorption sites¶
The substrate can be set using the setSubstrate method. Note that each instance of AdsorbedSurfaceGenerator can only set a substrate once. This is to avoid a mismatch between the substrate and adsorption sites, as well as the substrate and anchor atom site types and distributions. If a new substrate should be used, a new instance of AdsorbedSurfaceGenerator needs to be created. If the same molecules are to be used on the new substrate, use the copyMoleculeTypes method to copy any molecule types from the old instance to the new instance, i.e.
new_instance.copyMoleculeTypes(old_instance)
to copy all molecule types or
new_instance.copyMoleculeTypes(old_instance, 'water')
to copy only the molecule type ‘water’.
Note that this method does not copy the site_type or distribution from any anchor atoms, so these must be set in the new instance after copying to ensure that they match the new substrate and its adsorption sites. Similarly, if the parameters used to calculate adsorption sites in the old instance can be used again in the new instance, these can be retrieved using the adsorptionSiteParameters method, i.e.
new_instance.findAdsorptionSites(*old_instance.adsorptionSiteParameters())
Adsorption sites can be found using the findAdsorptionSites method. A list of basic adsorption site types can be found in Table 1.
The volume defined by the search_depth parameter should not contain atoms that are repeated in multiple layers, i.e. atoms should not have the same in-plane coordinates in multiple layers. Otherwise, undesired adsorption sites may be generated inside the substrate. See the figure below for clarification. The option to set the search_depth to an arbitrary value within the substrate thickness is useful for more complex crystal structures where adsorption sites are not all at the same height above the surface. This is especially useful for amorphous substrates, in which the surface volume is arbitrarily defined.

Fig. 163 Examples of the volume defined by surface_depth, a) with and b) without repeating atoms in multiple layers. In b), some of the red atoms are repeated in all three layers. Since the volume includes both the first and second layers, adsorption sites can be seen between these layers, inside the substrate.¶
The grouping_precision parameter can be used to further sort the basic adsorption site types by unique local environments. For instance, the short and long bridge sites on an elemental crystal FCC(110) surface can be sorted into ‘bridge_0’ and ‘bridge_1’.
Table 1. Basic adsorption site types. The substrate elements constituting the site are appended to the type in alphabetical order using an underscore. Additional sorting among each basic site type may be indicated by a trailing underscore and integer. Examples: ‘ontop_Pt_0’, ‘bridge_OPt_1’.
Site type |
Description |
|---|---|
‘ontop’ |
A surface site that is 1-fold coordinated. |
‘bridge’ |
A surface site that is 2-fold coordinated. |
‘hollow_3fold’ |
A surface site that is 3-fold coordinated. |
‘hollow_4fold’ |
A surface site that is 4-fold coordinated. Requires at least two layers of atoms in the substrate, otherwise these are labeled as bridge sites. |
‘hcp’ |
A 3-fold coordinated site that corresponds to hexagonal-close-packed stacking. Requires at least three layers of atoms in the substrate to be identified, otherwise these are labeled as ‘hollow_3fold’ sites. |
‘fcc’ |
A 3-fold coordinated site that corresponds to face-centered-cubic stacking. Requires at least three layers of atoms in the substrate to be identified, otherwise these are labeled as ‘hollow_3fold’ sites. |
Molecules and alignment¶
Molecule types can be added, modified and removed using the addMolecule, changeMolecule and removeMolecule methods, respectively.
Each molecule type will have one anchor atom by default, which cannot be removed. It is used to define how a molecule type should be distributed onto the substrate surface. Each anchor atom specifies the index of an atom in the molecule, the site type that that atom (equivalently, the molecule type) should occupy and how the molecule should be distributed on these sites.
For a single molecule (i.e.
count=1), the distribution may beRandomor a specific site.For multiple molecules, the distribution options are
RandomorQuasirandom.The
Quasirandomdistribution will attempt to space out the molecules among the sites, and approaches a uniform distribution for sufficiently large numbers of molecules.
The number of molecules of each molecule type can be set either as a coverage or a count. The coverage is calculated in terms of the number of sites that should be occupied for the chosen site type. If both count and coverage are given, the coverage will be used.
The molecule orientation can be given as an option from ORIENTATIONS; the available options are summarize in Table 2. The orientation can also be given as a set of custom rotation angles, by using the named tuple for which descriptions of the different angles are given in Table 3. Example:
ORIENTATION(surface_rotation_angle, surface_tilt_angle, molecule_rotation_angle)
If ORIENTATIONS.ANCHORED is used, it is expected that more than one anchor atom has been added. Additional anchor atoms can be added, modified and removed using the addAnchorAtom, changeAnchorAtom and removeAnchorAtom methods, respectively. If, however, additional anchor atoms have not been given but the orientation is set to ORIENTATIONS.ANCHORED, the generator will use the ORIENTATIONS.VERTICAL orientation. For additional anchor atoms beyond the first, the site type and distribution of each anchor atom should be set.
Similar to the first anchor atom, the distribution of additional anchor atoms can be set to a specific site of the chosen site type for a single molecule.
For both single and multiple molecules, the distribution of additional anchor atoms can also be set to
Random. In this case, this means the generator will find the site of the chosen site type that comes closest to the that anchor atom.
The same type of molecule can be added more than once. This can be useful when more precise control of the placement of each molecule of a single type is needed. To achieve this, add each molecule with a count of one and distribute it to a specific site type and site.
Table 2. Orientation options.
Orientation |
Description |
|---|---|
|
Orient the molecule such that it appears to stand vertically on the
substrate. This is the default orientation and corresponds to when all
rotation angles are equal to zero. Often, this orientation also
coincides with the |
|
Orient the molecule such that it appears to lay flat on the substrate. This corresponds to setting the tilt angle to 90 degrees. |
|
Orient the molecule such that the first (smallest) principle moment axis of inertia is aligned to the substrate surface normal. |
|
Orient the molecule such that the second principle moment axis of inertia is aligned to the substrate surface normal. |
|
Orient the molecule such that the third (largest) principle moment axis of inertia is aligned to the substrate surface normal. |
|
Orient the molecule by designating multiple anchor atoms. |
Table 3. Custom orientation angles. All angles are given in degrees.
Angle |
Range |
Description |
|---|---|---|
|
0 - 360 |
The angle of the molecule around the substrate surface normal. |
|
-90 - +90 |
The angle of the molecule towards the substrate surface,
in the direction or the |
|
0 - 360 |
The angle of the molecule around the axis defined by the
|