runPackmol¶
Included in QATK.Core
- runPackmol(molecules, cell_or_host, tolerance=None, buffer_size=None, maximum_loops=None, random_seed=None, terminal_output=False)¶
Run Packmol in a subprocess, interpret the output and return the result as a
BulkConfiguration.- Parameters:
molecules (list of (
MoleculeConfiguration, int)) – A list of tuples, (Molecules, Molecules count) to include in the packing.cell_or_host (
BravaisLattice|BulkConfiguration) – The packing volume or host configuration.tolerance (PhysicalQuantity of type length) – The target minimum intermolecular distance after packing. Corresponds to the tolerance keyword in packmol.
buffer_size (PhysicalQuantity of type length) – A buffer of this size is added to the cell vectors after packing to facilitate periodic boundary conditions.
maximum_loops (int) – The maximum number of loops in the packing optimization. Corresponds to the nloop keyword in packmol.
random_seed (int | None | Random) – The random seed to give Packmol. Use None to generate a seed automatically. Use Random to generate a different seed with each call.
terminal_output (bool) – If True, print Packmol message to the terminal.
- Returns:
Tuple containing the packed configuration, the success flag from reading the Packmol output and the Packmol output message.
- Return type:
tuple(
BulkConfiguration, bool, str)
Usage Examples¶
The first example demonstrates the packing of 10 CH4 molecules together with 40 H2O in a cube with side-length 40 Angstrom, using a maximum of 30 packing loops, a tolerance of 2 Angstrom and a buffer region size, also of 2 Angstrom (see Notes for further details on these input parameters):
# --------------------------------------------------------------------------
# Methane
# --------------------------------------------------------------------------
# Define elements
elements = [Carbon, Hydrogen, Hydrogen, Hydrogen, Hydrogen]
# Define coordinates
coordinates = [[ -3.67767153e-08, -5.21170217e-07, -1.77696852e-05],
[ 6.30525881e-01, -8.91103633e-01, 2.66535938e-02],
[ -6.29878738e-01, 2.68953440e-02, 8.91606540e-01],
[ -6.30920686e-01, -2.70480029e-02, -8.90758789e-01],
[ 6.30273980e-01, 8.91262502e-01, -2.72895999e-02]] * Angstrom
# Set up configuration
methane = MoleculeConfiguration(
elements,
coordinates
)
# --------------------------------------------------------------------------
# H2O
# --------------------------------------------------------------------------
# Define elements
elements = [Oxygen, Hydrogen, Hydrogen]
# Define coordinates
coordinates = [[ 0.0, 4.0, 0.0],
[ 0.76923955, 4.0, -0.59357141],
[ -0.76923955, 4.0, -0.59357141]] * Angstrom
# Set up configuration
water = MoleculeConfiguration(
elements,
coordinates
)
# Create a list of molecules and their numbers
molecules = [
(methane, 10),
(water, 40)
]
# Define the volume in which the molecules should be packed
u1, u2, u3 = numpy.diag([40.0] * 3) * Angstrom
cell = UnitCell(u1, u2, u3)
# Set the size of the buffer region
buffer_size = 2.0 * Angstrom
# Set Packmol parameters
tolerance = 2.0 * Angstrom
maximum_loops = 30
# Run the Packmol script to generate the packed configuration
packed_configuration, packing_successful, packmol_message = runPackmol(
molecules,
cell,
tolerance,
buffer_size,
maximum_loops,
)
print(f'Packing successful: {packing_successful}')
print(f'Packmol message: {packmol_message}')
print(f'Returned configuration: {packed_configuration}')
runPackmol_molecules_example.py
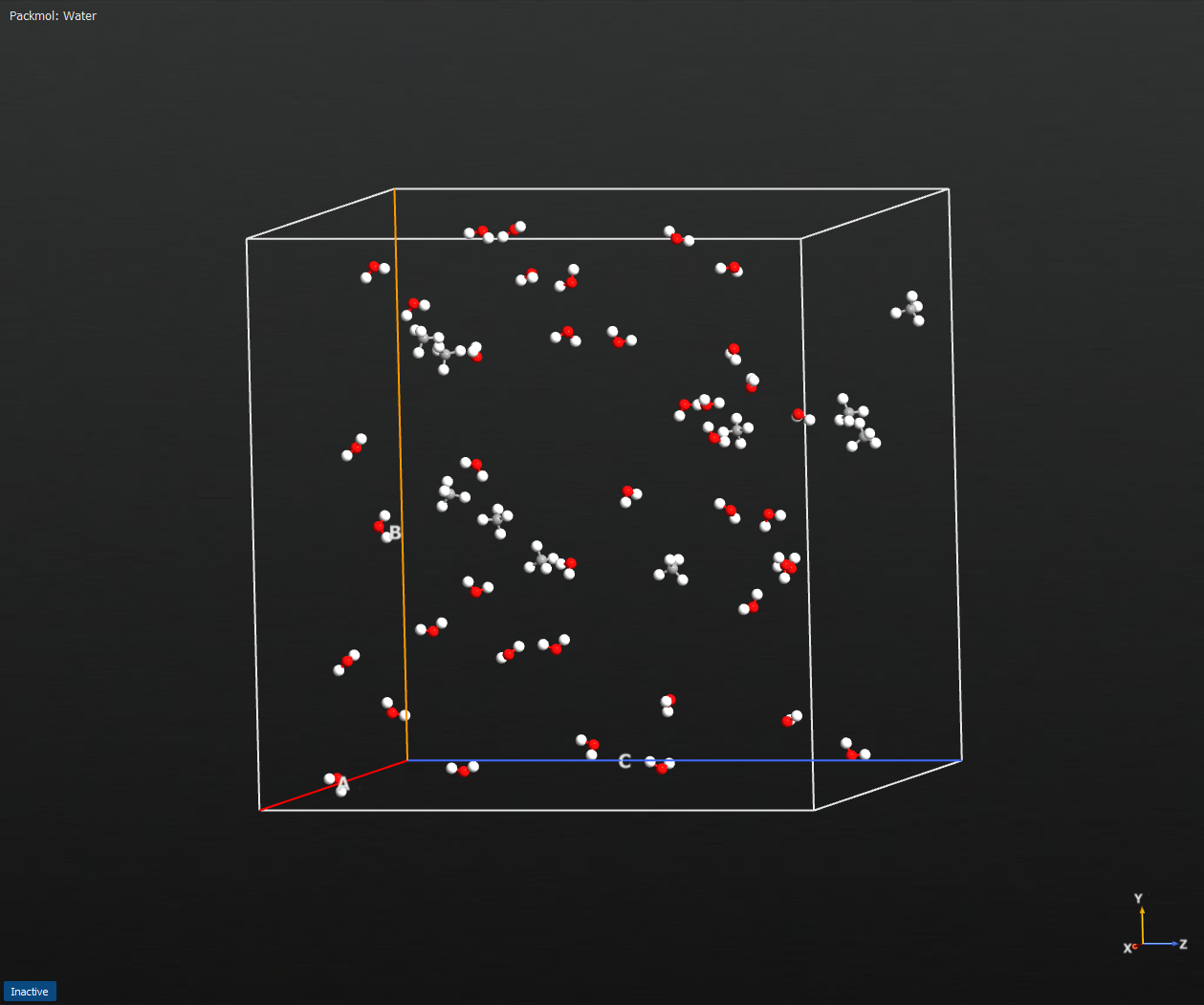
Fig. 209 Example output configuration from runPackmol, where the red-white molecules are H2O and the grey-white molecules are CH4.¶
In the code above, the cell is defined as a Unitcell, though any valid
BravaisLattice will work, also those that are non-orthogonal. As an example, try
replacing the lines
# Define the volume in which the molecules should be packed
u1, u2, u3 = numpy.diag([40.0] * 3) * Angstrom
cell = UnitCell(u1, u2, u3)
with
# Define the volume in which the molecules should be packed
cell = Triclinic(14.0 * Angstrom, 17.9999 * Angstrom, 22.0 * Angstrom,
45.0 * Degrees, 90.0 * Degrees, 90.0 * Degrees)
The second example demonstrates the packing of four CH4 into a zeolite host configuration (loaded from zeolite_LIT.py, which can be found as a separate download below), using a maximum of 30 packing loops, a tolerance of 2 Angstrom and a buffer region size also of 2 Angstrom:
# Import the host configuration
from zeolite_LIT import zeolite_LIT
# --------------------------------------------------------------------------
# Methane
# --------------------------------------------------------------------------
# Define elements
elements = [Carbon, Hydrogen, Hydrogen, Hydrogen, Hydrogen]
# Define coordinates
coordinates = [[ -3.67767153e-08, -5.21170217e-07, -1.77696852e-05],
[ 6.30525881e-01, -8.91103633e-01, 2.66535938e-02],
[ -6.29878738e-01, 2.68953440e-02, 8.91606540e-01],
[ -6.30920686e-01, -2.70480029e-02, -8.90758789e-01],
[ 6.30273980e-01, 8.91262502e-01, -2.72895999e-02]] * Angstrom
# Set up configuration
methane = MoleculeConfiguration(
elements,
coordinates
)
# Create a list of molecules and their numbers
molecules = [(methane, 4)]
# Set the size of the buffer region
buffer_size = 2.0 * Angstrom
# Set Packmol parameters
tolerance = 2.0 * Angstrom
maximum_loops = 30
# Run the Packmol script to generate the packed configuration
packed_configuration, packing_successful, packmol_message = runPackmol(
molecules,
zeolite_LIT,
tolerance,
buffer_size,
maximum_loops,
)
print(f'packing successful: {packing_successful}')
print(f'packmol message: {packmol_message}')
print(f'Returned configuration: {packed_configuration}')
runPackmol_molecules_with_host_example.py
zeolite_LIT.py
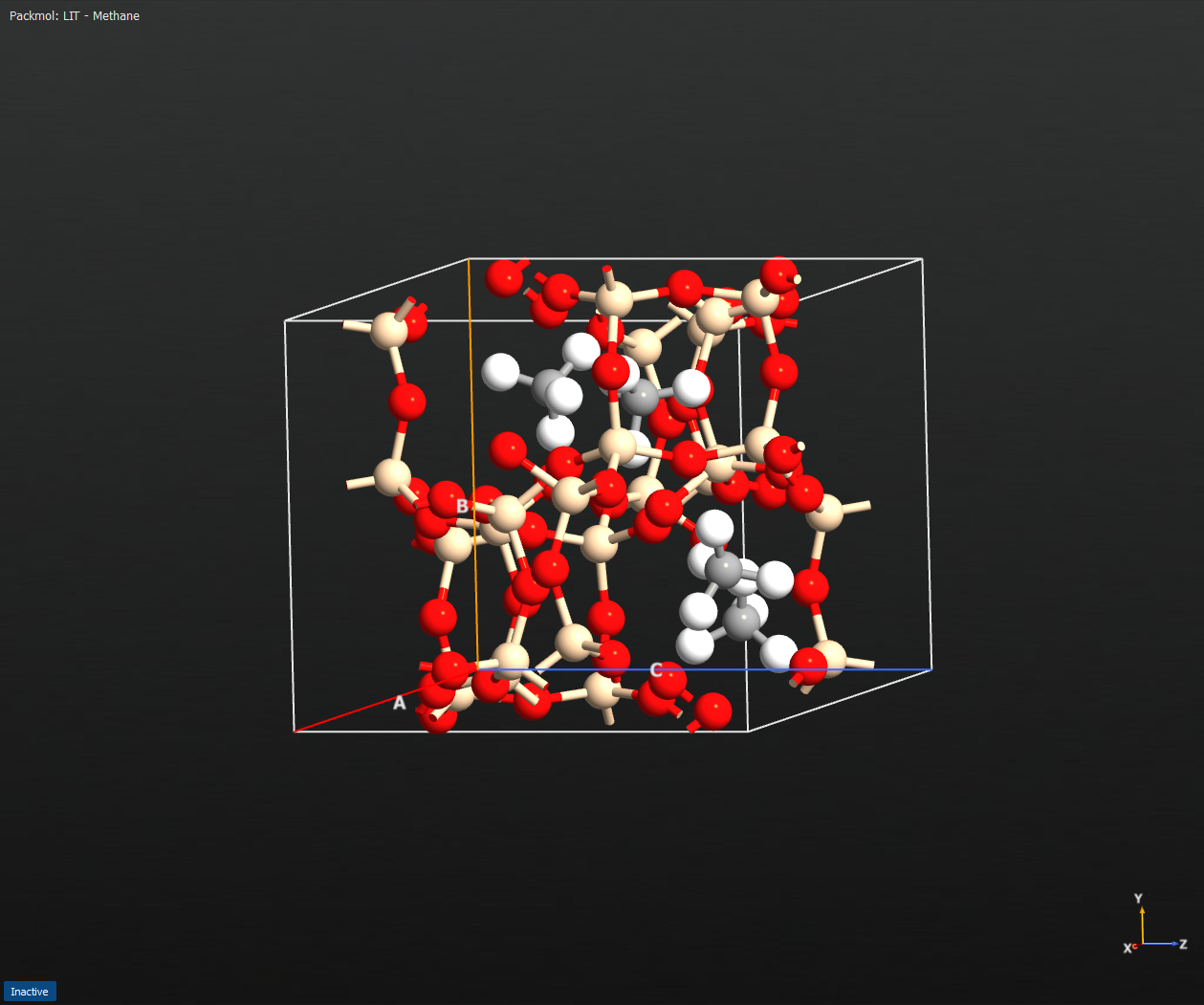
Fig. 210 Example output configuration from runPackmol, where the grey-white molecules are CH4. and the surrounding atoms in red and tan belong to the host configuration.¶
General¶
Packmol is a utility used in QuantumATK to generate configurations of packed molecules.
Normally, Packmol is available through the Packmol Builder Plugin.
The function runPackmol is meant to be an easy way to add Packmol functionality to a script written within the QuantumATK framework. It takes the same inputs as the Packmol Builder Plugin, which are:
Arbitrary types and numbers of molecules to be packed.
The cell into which the molecules should be packed, as a
BravaisLatticeor a host configuration into which the molecules should be packed.Packing density tolerances, i.e. the minimum acceptable distance between molecules.
A buffer size to avoid faulty packing at periphery of the cell. (See Notes for details)
The maximum number of optimization loops performed by Packmol.
For the original article presenting Packmol, see [1].
The official homepage of Packmol is (as of 2019) http://m3g.iqm.unicamp.br/packmol/home.shtml
Notes¶
Molecules must be given as a list of tuples, in which each tuple contains a pair of values: a molecule and the number of this molecule.
Natively, Packmol does not consider packing tolerances across the boundaries of the packing volume (i.e. it does not support periodic boundary conditions). To overcome this, the original authors suggest to use a buffer region around the packing volume which is subtracted from the total packing volume. This region will be left empty after the packing, and it is suggested to use energy minimization and equilibration on the packing volume with mirror images as a follow-up procedure to remove the empty spaces and complete the packing. In general, a buffer_size of 2 Angstroms for a cell length of 100 Angstroms is sufficient, though this may need to be increased for large molecules.
In general, a tolerance of 2 Angstroms is sufficient. Special cases may arise for cyclic systems such as toluene, which may become linked during packing. In such cases, a larger tolerance is more suitable.