DeviceConfiguration¶
Included in QATK.Core
- class DeviceConfiguration(central_region, electrodes, equivalent_electrode_lengths=None, transverse_electrode_repetitions=None)¶
A two-probe configuration consisting of a central region coupled to two electrodes.
- Parameters:
central_region (
BulkConfiguration) – The scattering region of the device.electrodes (sequence (size 2) of
BulkConfiguration) – The semi-infinite regions on either side of the device.equivalent_electrode_lengths (sequence (size 2) of PhysicalQuantity of type length) – The lengths to use for the equivalent electrode regions in the central region. Default: Length of the actual electrodes given.
transverse_electrode_repetitions (sequence (size 2) of sequence (size 2) of int) – Repetitions of the electrodes in the A and B directions, given as a list of two integers for both electrodes. Default:
[[1, 1], [1, 1]]
- addBonds(bond_list)¶
Add bonds.
- Parameters:
bond_list (numpy.ndarray) – The list of bonds to add.
- addTags(tags, indices=None)¶
Add a set of tags to atoms matching a collection of indices.
- Parameters:
tags (list | str) – The list of tags to add to matching atoms.
indices (list | int | None) – The list of indices to match atoms against. Default: All indices.
- atomicMasses()¶
- Returns:
The masses of the atoms in the configuration.
- Return type:
PhysicalQuantity of type mass
- atomicNumbers()¶
- Returns:
The list of atomic numbers associated with the elements.
- Return type:
list of ints
- bias()¶
Calculate the bias of the attached calculator If no calculator is attached return None.
- Returns:
The bias.
- Return type:
PhysicalQuantity with electric potential units | None
- bonds()¶
- Returns:
An array with the the two atom indices for each bond in the central region along with the vector which periodic images this bond connects.
- Return type:
array
- bravaisLattice()¶
- Returns:
The bravais lattice of the central region.
- Return type:
- calculator()¶
- Returns:
The calculator attached to the configuration, i.e. the calculator that will be used for both simulation and analysis.
- Return type:
- cartesianCoordinates()¶
The Cartesian coordinates of the atoms in the the central region of the configuration.
- Returns:
The Cartesian coordinates.
- Return type:
PhysicalQuantity of type length
- centralRegion()¶
The central region of the device.
- Returns:
The central region.
- Return type:
- coarseGrainDescriptors()¶
- Returns:
The list of either elements or coarse grain particles for each element. Isotopes are returned as just their base element, UnitedAtoms and Particle are given as their specific type.
- Return type:
list of type ParticleDescriptor or PeriodicTableElement
- copy()¶
- Returns:
A copy of the current configuration.
- Return type:
MoleculeConfiguration|BulkConfiguration|DeviceConfiguration|SurfaceConfiguration
- copyAndDeleteAtoms(indices)¶
Create a new configuration by deleting some atoms from this configuration.
- Parameters:
indices (list of int) – The indices of the atoms to delete.
- Returns:
The configuration with some atoms deleted.
- Return type:
MoleculeConfiguration|BulkConfiguration|DeviceConfiguration|SurfaceConfiguration
- copyAndMerge(other)¶
Create a new configuration by merging this configuration with another configuration.
- Parameters:
other (
MoleculeConfiguration|BulkConfiguration|DeviceConfiguration|SurfaceConfiguration) – The other configuration.- Returns:
The merged configuration.
- Return type:
MoleculeConfiguration|BulkConfiguration|DeviceConfiguration|SurfaceConfiguration
- copyAndShiftAtoms(displacement, indices=None)¶
Create a new configuration with some atoms translated.
- Parameters:
displacement (PhysicalQuantity of type length) – The displacement that should be applied to the atom positions.
indices (list of int) – The indices to shift. Default: All.
- Returns:
The configuration with the translation applied.
- Return type:
MoleculeConfiguration|BulkConfiguration|DeviceConfiguration|SurfaceConfiguration
- crossSection()¶
Return the cross-sectional area of the central region.
- Returns:
The cross-sectional area.
- Return type:
float
- deleteAtoms(indices)¶
Delete the specified atoms.
- Parameters:
indices (array of int) – The indices to delete.
- deleteBonds(bond_list=None, pair_selection=None)¶
Delete bonds connected to atomic indices.
- Parameters:
bond_list (A two-dimensional sequence) – The pairs of bondes indices.
pair_selection (list | None) – Specifies two groups between which bonds are delete. Selectable groups are elements, index lists, tag names, or None (all atoms).
- dielectricRegions()¶
- Returns:
The dielectric regions in the central region.
- Return type:
list of
BoxRegion|SphereRegion|TubeRegion
- electrodes()¶
The electrodes of the device.
- Returns:
The pair of electrodes belonging to this configuration.
- Return type:
list of
BulkConfiguration
- electrodesDisplacement()¶
The displacements of the
BravaisLatticeof the electrodes in the C-direction in order to match theBravaisLatticeof the central_region.- Returns:
The electrode displacements.
- Return type:
PhysicalQuantity of type length
- electrodesWithCalculator()¶
- Returns:
A copy of the electrodes of the configuration with attached electrode calculator. If the configuration is updated, the calculators include a copy of the updated state.
- Return type:
list of
BulkConfiguration
- elements()¶
- Returns:
The elements in configuration.
- Return type:
list of
PeriodicTableElement
- externalPotential()¶
- Returns:
The external potential present in the central region.
- Return type:
- findBonds(fuzz_factor=1.1, pair_selection=None)¶
Find bonds in the configuration according to the combined covalent radii of the element pairs, multiplied with a fuzz factor. Optionally, find bonds only between two specified sub-groups of atoms. The bonds are primarily used in to set the topology of bonded potentials in the TremoloX-calculator.
- Parameters:
fuzz_factor (float) – The factor by which the covalent radii are multiplied to determine the cutoff distance for a bond.
pair_selection (list(2) of type PeriodicTableElement, list of int, or str.) – Specifies two groups between which bonds are detected. Selectable groups are elements, index lists, tag names, or None (all atoms). By default bonds between all atoms in the configuration are taken into account.
- fixedSpinDirections()¶
- Returns:
The fixed spin directions for the configuration.
- Return type:
FixedSpin| None
- fractionalCoordinates()¶
- Returns:
The fractional coordinates of the central region.
- Return type:
array of floats
- generateShifts()¶
Method for generating a list of origin shifts along two lattice directions.
It will create a list of 9 shifts of the supercell origin (-1 to 1) in x and y periodic directions.
- Returns:
The array of shifts.
- Return type:
PhysicalQuantityof type length
- ghostAtoms()¶
- Returns:
The list of ghost atoms of the central region.
- Return type:
list of ints
- improperDihedralIndices()¶
- Returns:
The list of atom indices for each improper dihedral or None if no improper dihedrals are defined. Improper dihedrals are mainly used in bonded force fields.
- Return type:
numpy array | None
- indices()¶
- Returns:
The indices of the atoms.
- Return type:
list of int
- indicesFromIsotopes(isotopes)¶
- Parameters:
isotopes (list of type PeriodicTableElement or Isotope) – The isotopes to select.
- Returns:
The indices of the selected isotopes.
- Return type:
list of type int
- indicesFromTags(tags=None)¶
List the indices associated with a given collection of tags.
- Parameters:
tags (list | str) – A list of tags for which all matching indices should be extracted.
- Returns:
The list of indices corresponding to the specified tag name(s).
- Return type:
list of ints
- magneticMoments()¶
- Returns:
The magnetic moments of the atoms. Has the dimensionality nx3.
- Return type:
PhysicalQuantity of type Bohr magneton.
- merge(other)¶
Merge configuration with another configuration.
NOTE: For devices this method shifts the atoms of the other configuration to the center.
- Parameters:
other (AtomicConfiguration) – A different AtomicConfiguration.
- metallicRegions()¶
- Returns:
The metallic regions in the central region.
- Return type:
list of
BoxRegion|SphereRegion|TubeRegion
- metatext()¶
- Returns:
The metatext of the object or None if no metatext is present.
- Return type:
str | None
- nlinfo()¶
- Returns:
The configuration information.
- Return type:
dict
- nlprint(stream=None)¶
Print a string containing an ASCII table useful for plotting the AtomicConfiguration object.
- Parameters:
stream (python stream) – The stream the table should be written to. Default:
NLPrintLogger().
- numberOfAtoms()¶
- Returns:
The total number of atoms in the configuration.
- Return type:
int
- partialCharges(indices=None)¶
Get the list of partial atomic charges that can be used for representing electrostatic interactions in ATK-ForceField.
- Parameters:
indices (list | int | None) – The indices for which to return the partial charges. Default: All indices.
- Returns:
A PhysicalQuantity array of the atomic partial charge for each atom.
- Return type:
PhysicalQuantity of type charge | None
- particleDescriptors()¶
- Returns:
The list of particle descriptors for each atom. This should return the exact same list as was given in the constructor argument “elements”.
- Return type:
list of type ParticleDescriptor or PeriodicTableElement
- static periodicBoundaries()¶
- Returns:
The periodic boundary conditions of the configuration.
- Return type:
list
- primitiveVectors()¶
- Returns:
The primitive lattice vectors.
- Return type:
PhysicalQuantity of type length
- removeTags(tags=None, indices=None, purge=False)¶
Remove a set of tags from atoms matching a collection of indices.
- Parameters:
tags (list | str) – The list of tags to add to matching atoms. Default: All tags.
indices (list | int) – The list of indices to match atoms against. Default: All indices.
purge (bool) – When removing tags from the configuration, delete the tag completely when not associated with any atoms anymore. Default: False
- repeat(na=1, nb=1, nc=1, stack_systems=False)¶
Repeat the derived class with the integer values na, nb, and nc along the three primitive unit cell vectors. The repeated system is constructed with a cell of the type UnitCell.
- Parameters:
na (int) – The repetition along the a-axis. Default: 1.
nb (int) – The repetition along the b-axis. Default: 1.
nc (int) – The repetition along the c-axis. Default: 1.
stack_systems (bool) – If True the basis atoms are repeated as a unit, i.e. a0 and b0 are repeated as: a0,b0,a1,b1, … If False the basis atom are repeated individually, i.e. a0 and b0 are repeated as: a0,a1,…, b0,b1,… Default: True.
- Returns:
The repeated system.
- Return type:
- scalePartialCharges(scale_factor, indices=None)¶
Scale the partial charges with a given scale factor. These partial charges are used with Forcefield calculators.
- Parameters:
scale_factor (float) – The factor for scaling charges.
indices (list | int | None) – The indices for which to set the total charge. Default: All indices.
- setBonds(bond_list, skip_checks=False)¶
Set the bonds on the configuration. The bonds are primarily used in to set the topology of bonded potentials in the TremoloX-calculator.
- Parameters:
bond_list (list(n, 2) | list(n, 5) | None) – A list which contains for each bond the indices of the two connected atoms. Optionally, three more integers can be specified for each bond, which must be between -1 and 1, and which denote to which neighboring image cell the bond is connected. Without these additional indices, the minimum image convention is obeyed.
skip_checks (bool) – Skip argument type checking and just directly assign the value.
- setCalculator(calculator, initial_state=None, initial_spin=None)¶
Attach a Calculator to the configuration which will be used in calculations involving the configuration.
- Parameters:
calculator (Calculator) – The calculator object that should be attached to the configuration.
initial_state (
DeviceConfigurationwith a calculator | None) – The initial state to be used for this configuration. Default: No initial state.initial_spin (
InitialSpin| None) – The initial InitialSpin object to be used for this configuration. Default:InitialSpin(), except when used with a calculator of typeHuckelCalculator,SlaterKosterCalculator,SemiEmpiricalCalculator,DeviceHuckelCalculator,DeviceSlaterKosterCalculator,DeviceSemiEmpiricalCalculator. In this caseInitialSpin([0., 0., ...])is used, i.e. the scaled spins for each atom are set to 0.
- setCartesianCoordinates(cartesian_coordinates, indices=None, skip_checks=False)¶
Set the Cartesian coordinates of the atoms in the central region. Changes in the coordinates of the electrode extension will be synchronized with the corresponding coordinates in the electrodes.
- Parameters:
cartesian_coordinates (PhysicalQuantity of type length) – The new coordinates of the atoms in each image.
indices (list) – The indices of the atoms to set the positions of. Default: All indices.
skip_checks (bool) – Skip argument type checking and just directly assign the value. Default: False
- setDielectricRegions(dielectric_regions)¶
Set the dielectric regions for the central region.
- Parameters:
dielectric_regions (list of
BoxRegion|SphereRegion|TubeRegion) – The list of dielectric regions to set.
- setExternalPotential(external_potential)¶
Set an external potential on the configuration that will be used in calculations involving the configuration.
- Parameters:
external_potential (
AtomicShift|AtomicCompensationCharge) – The external potential to apply.
- setImproperDihedralIndices(improper_dihedral_indices)¶
Set the list of atom indices for each improper dihedral in bonded force fields.
- Parameters:
improper_dihedral_indices (list or array with shape (m, 4) | None) – The list of the 4 indices defining the connectivity for each improper dihedral or None to delete the current dihedral connectivity.
- setMagneticField(magnetic_field)¶
Set local magnetic field. The spins will be forced to point in the directions given by the magnetic_field object. The magnetic field can be defined for each atom. This only has an effect for Noncollinear or Spinorbit calculations.
- Parameters:
magnetic_field (
FixedSpin) – The magnetic field to be applied.
- setMagneticMoments(magnetic_moments=None, skip_checks=False)¶
Function to set magnetic moments on the configuration.
- Parameters:
magnetic_moments (PhysicalQuantity of type Bohr magneton.) – The magnetic_moments to set on the configuration. Has the dimensionality nx3. Default: None.
skip_checks (bool) – Skip argument type checking and just directly assign the value. Default: False.
- setMetallicRegions(metallic_regions)¶
Set the metallic regions for the central region.
- Parameters:
metallic_regions (list of
BoxRegion|SphereRegion|TubeRegion) – The list of metallic regions to set.
- setMetatext(metatext)¶
Set a given metatext string on the object.
- Parameters:
metatext (str | None) – The metatext string to set. Use “None” to remove current metatext.
- setPartialCharges(charge_list, indices=None, skip_checks=False, update_calculator=True)¶
Set the partial charges on the configuration. The partial charges are used primarily to model electrostatic interactions in the TremoloX-calculator.
- Parameters:
charge_list (PhysicalQuantity of type charge | None) – A list of atomic partial charges which contains a charge for each atom.
indices (list | int | None) – The indices for which to set the partial charges. Default: All indices.
skip_checks (bool) – Skip argument type checking and just directly assign the value. Default: False.
update_calculator (bool) – Whether to update and attached Forcefield calculator with the new charges. Default: True.
- setVelocities(velocities=None, skip_checks=False)¶
Function to set velocities on the configuration.
- Parameters:
velocities (PhysicalQuantity of type velocity | None) – The velocities to set on the configuration. Has the dimensionality nx3. Default: None.
skip_checks (bool) – Skip argument type checking and just directly assign the value. Default: False.
- shiftAtoms(displacement, indices=None, skip_checks=False)¶
Translate some atoms.
- Parameters:
displacement (PhysicalQuantity of type length) – The displacement that should be applied to the atom positions.
indices (list of int) – The indices to shift. Default: All.
skip_checks (bool) – True, if all consistency checks should be skipped.
- shiftPartialCharges(total_charge, indices=None)¶
Shift the partial charges so that their sum is the given total charge value. These partial charges are used with Forcefield calculators.
- Parameters:
total_charge (PhysicalQuantity of type charge) – The new total charge.
indices (list | int | None) – The indices for which to set the total charge. Default: All indices.
- symbols()¶
- Returns:
The element symbols of the configuration.
- Return type:
list of str
- tags(indices=None)¶
List the tags associated with a given collection of indices. The list returned is the set union of tags associated with the given indices. If no collection of indices is provided, then all tags on the configuration are returned.
- Parameters:
indices (list | int) – The indices to check. Default: All indices.
- Returns:
The set union of tags present on the provided indices.
- Return type:
set
- transverseElectrodeRepetitions()¶
- Returns:
The transverse electrode repetitions.
- Return type:
list (size 2) of list (size 2) of int
- uniqueElements(ordered=False)¶
- Parameters:
ordered (bool) – If the elements should be returned in ascending order by atomic number.
- Returns:
The unique elements contained in the configuration.
- Return type:
list of
PeriodicTableElement
- uniqueString()¶
Return a unique string representing the state of the object.
- update(force_restart=False)¶
A self-consistent solution is generated, using the currently set calculator.
- Parameters:
force_restart (bool) – Force the self-consistent calculation to restart. Default: False.
- velocities()¶
- Returns:
The velocities of the atoms. Has the dimensionality nx3.
- Return type:
PhysicalQuantity of type velocity
Usage Examples¶
Construct a device system consisting of a hydrogen molecule connected with two chains of Lithium atoms:
# Define A,B directions of lattice
vector_a = [5.0, 0.0, 0.0]*Angstrom
vector_b = [0.0, 5.0, 0.0]*Angstrom
# setup electrode
electrode = BulkConfiguration(
bravais_lattice=UnitCell(vector_a, vector_b,
[0.0, 0.0, 9.0]*Angstrom),
elements=[Lithium, Lithium, Lithium],
cartesian_coordinates=[[ 2.5, 2.5, 1.5],
[ 2.5, 2.5, 4.5],
[ 2.5, 2.5, 7.5]]*Angstrom
)
# setup Central region
central_region = BulkConfiguration(
bravais_lattice= UnitCell(vector_a, vector_b,
[0.0, 0.0, 22.0]*Angstrom),
elements=[Lithium, Lithium, Lithium, Hydrogen, Hydrogen,
Lithium, Lithium, Lithium],
cartesian_coordinates=[[ 2.5, 2.5, 1.5],
[ 2.5, 2.5, 4.5],
[ 2.5, 2.5, 7.5],
[ 2.5, 2.5, 10.5],
[ 2.5, 2.5, 11.5],
[ 2.5, 2.5, 14.5],
[ 2.5, 2.5, 17.5],
[ 2.5, 2.5, 20.5]]*Angstrom
)
#setup Device configuration
device_configuration = DeviceConfiguration(
central_region,
[electrode, electrode]
)
Construct a device system from bulk lithium with transverse electrode repetitions:
# Set up the left minimal electrode with 2 Li atoms.
left_electrode_lattice = Hexagonal(3.111*Angstrom, 5.093*Angstrom)
left_electrode_elements = [Lithium, Lithium]
left_electrode_coordinates = [[ 0.333333333333, 0.666666666667, 0.25 ],
[ 0.666666666667, 0.333333333333, 0.75 ]]
left_electrode = BulkConfiguration(
bravais_lattice=left_electrode_lattice,
elements=left_electrode_elements,
fractional_coordinates=left_electrode_coordinates,
)
# Use the same electrode as the right electrode.
right_electrode = left_electrode
# Define transverse electrode repetitions; 2 repetitions
# in A and 3 in B for both electrodes.
transverse_electrode_repetitions = [[2, 3], [2, 3]]
# Create the repeated electrode and repeat it 4 times along
# C to create the central region. In this manner, the atoms in the electrode
# extensions will match the repeated electrodes.
repeated_left_electrode = left_electrode.repeat(
transverse_electrode_repetitions[0][0],
transverse_electrode_repetitions[0][1],
1)
central_region = repeated_left_electrode.repeat(1, 1, 4)
# Change one of the atoms at the center of the central region to make a non-homogeneous device.
central_region_lattice = central_region.bravaisLattice()
central_region_elements = central_region.elements()
central_region_coordinates = central_region.fractionalCoordinates()
central_region_elements[24] = Hydrogen
central_region = BulkConfiguration(
bravais_lattice=central_region_lattice,
elements=central_region_elements,
fractional_coordinates=central_region_coordinates,
)
# Set up the device configuration with transverse repetitions.
device_configuration = DeviceConfiguration(
central_region=central_region,
electrodes=[left_electrode, right_electrode],
transverse_electrode_repetitions=transverse_electrode_repetitions,
)
Notes¶
ATK recognizes four types of atomic geometries:
Molecules (MoleculeConfiguration)
Bulk (BulkConfiguration)
One Probe (SurfaceConfiguration)
Two Probe (DeviceConfiguration)
A two-probe setup is created by attaching two bulk electrodes to the left and the right of a central region. The properties within the electrode regions are fixed at their bulk properties, whereas the central region is described self-consistently. The device configuration must fulfill the following properties:
The electrodes and the central BulkConfiguration objects must have the same cell vectors in the A and B direction. If
transverse_electrode_repetitionshave been specified, the electrodes are assumed to be the minimal, irreducible ones in the A and B directions. In this case, the electrode unit cell vectors in the A and B directions multiplied by the transverse electrode repetitions must equal the central region’s lattice vectors.The C direction must be perpendicular to the A and B directions.
The first atoms in the central region must be positioned identically to the atoms in the left electrode (repeated by the correponding repetitions in A and B if
transverse_electrode_repetitionshave been specified) when the central region AB faces are aligned with the AB faces of the electrodes. See Fig. 169 for an example.Similarly, the last atoms in the central region must be positioned identically to the atoms in the right (repeated) electrode. See Fig. 169 for an example.
The parameter
equivalent_electrode_lengthscan be used to control the number of electrode repeats along the C direction in the electrode extensions of the central region. The sum of the left and right equivalent electrode lenghts cannot exceed the lenght of the central region. Fig. 169 shows an example of two repeats of both the left and right electrodes along C in the corresponding electrode extensions. If the left and right electrode lengths are 5 and 7Angstrom, respectively, this example corresponds to setting the parameter to the valueequivalent_electrode_lengths=[2 * 5.0, 2 * 7.0] * Angstrom.
If there is structural periodicity in the electrodes in the A and/or B directions, using the
minimal, irreducible electrodes and specifying the corresponding
transverse_electrode_repetitions can speed up the device calculation significantly and reduce
memory usage.
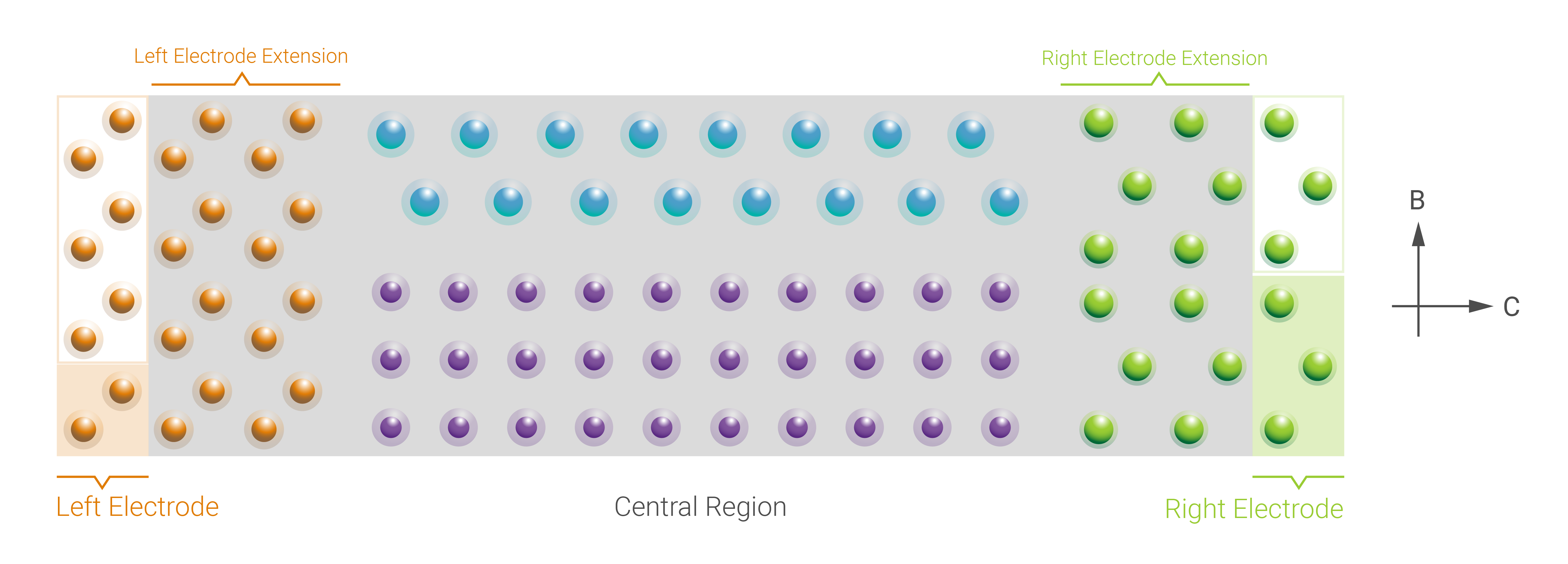
Fig. 169 Example of the constituents of a device system with 2 electrodes. The constituents are the left
electrode, central region, and right electrode. The central region of the device is marked with a
gray color, and the electrode extension regions of the central region are also shown. In this
case, the repeated left electrode consists of four repetitions of the minimal electrode along the
B direction, whereas the repeated right electrode consists of two repetitions along B of the
corresponding minimal electrode, i.e., transverse_electrode_repetitions=[[1, 4], [1, 2]]
(note that the A direction is not shown, and assumed not to have repetitions). Furthermore, the
equivalent electrode lenghts in the central region correspond to twice the length of the
electrodes in the C direction.¶
Attention
If transverse_electrode_repetitions have been specified, the k_point_sampling of the
electrode_calculators in the device calculator (see DeviceLCAOCalculator and
DeviceSemiEmpiricalCalculator) must be compatible with the k_point_sampling of the
central region, i.e., the k-point density of the minimal electrodes along the A and B directions
must be the same as the k-point density in the central region along A and B.
Vacuum basis sets (ghost atoms) can be included in the DeviceConfiguration by specifying a
ghost_atoms list for the BulkConfiguration of the central and electrode regions.