MDTrajectory¶
Included in QATK.Dynamics
- class MDTrajectory(path=None, group_name=None, overwrite=None)¶
Constructor for the MDTrajectory that contains the configurations from a
MolecularDynamics()run.- Parameters:
path (str | None) – The path to the HDF5 file. If the path is None then all data will be stored in memory and no file will be written. Default: None
group_name (str | None) – The name of the HDF5 group to store the data in. If the value is None then a new group will be automatically made. Default: None
overwrite (bool) – If not False, any existing hdf5 group named group_name will be overwritten. Default: False
- calculator()¶
- Returns:
The calculator if one is available, otherwise None.
- Return type:
Calculator | None
- cells(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The cell vectors of the images or None if the trajectory length is zero or the configuration is a MoleculeConfiguration.
- Return type:
PhysicalQuantity of type length | None
- concatenate(*trajectories, interval=1, path=None, object_id=None)¶
Combine several MDTrajectory objects into a new MDTrajectory.
- Parameters:
trajectories (list of MDTrajectory objects) – List of MDTrajectory objects to concatenate to this trajectory. The trajectories must be continuous, i.e. the last images of the previous trajectory must match the first image of the next trajectory.
interval (int) – Only keep every interval steps.
path (str | None) – The path to the HDF5 file. If the path is None then all data will be stored in memory and no file will be written. Default: None
object_id – The object id to use. If the value is None then an id will be automatically made. Default: None
- Returns:
The concatenated MDTrajectory.
- Return type:
- coordinates(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The Cartesian coordinates of the images or None the trajectory length is zero.
- Return type:
PhysicalQuantity of type length | None
- elements()¶
- Returns:
The elements of the images as a list of length n or None if the trajectory length is zero.
- Return type:
list of type
PeriodicTableElement
- forces(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The forces of the images. If the trajectory length is zero or the forces were not written during the MolecularDynamics run then None is returned.
- Return type:
PhysicalQuantity of type energy/length | None
- image(image_index, set_calculator=True, set_velocities=True, recycle_configuration=False)¶
- Parameters:
image_index (int) – The index of the configuration that should be extracted.
set_calculator (bool) – If True, the calculator is set on the returned image. This can be disabled to increase the performance when iterating over images, because the overhead of copying and setting up a new calculator can be large. Default: True
set_velocities (bool) – If True, the velocities are set on the returned image. This can be disabled to increase performance. Default: True
recycle_configuration (bool) – If True, the same configuration instance (with updated coordinates/cell) is returned each time image is called. This can increase performance, when iterating through images, but only allows for one configuration to exist at a time. Default: False
- Returns:
A copy of the configuration with the image_index.
- Return type:
MoleculeConfiguration|BulkConfiguration|DeviceConfiguration|SurfaceConfiguration
- images(set_calculator=True, set_velocities=True)¶
- Parameters:
set_calculator (bool) – If True, the calculator is set on the returned image. This can be disabled to increase the performance when iterating over images, because the overhead of copying and setting up a new calculator can be large. Default: True
set_velocities (bool) – If True, the velocities are set on the returned image. This can be disabled to increase performance. Default: True
- Returns:
All the configurations in the trajectory.
- Return type:
list of
MoleculeConfiguration|BulkConfiguration|DeviceConfiguration|SurfaceConfiguration
- kineticEnergies(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The kinetic energies of the images or None the trajectory length is zero.
- Return type:
PhysicalQuantity of type energy | None
- lastImage()¶
- Returns:
The last image in the trajectory with the calculator attached.
- Return type:
MoleculeConfiguration|BulkConfiguration|DeviceConfiguration|SurfaceConfiguration
- lastStep()¶
- Returns:
The step number of the last step.
- Return type:
int
- lastTime()¶
- Returns:
The time associated with the last step.
- Return type:
PhysicalQuantity of type time
- length()¶
- Returns:
The number of images in the trajectory.
- Return type:
int
- masses()¶
- Returns:
The masses of the atoms with shape (num_atoms,) or None if the trajectory length is zero.
- Return type:
PhysicalQuantity of type mass
- measurement(name, idx=Ellipsis)¶
- Parameters:
name (str) – The name of the measurement.
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
A 2-tuple of the time and the measurement.
- Return type:
tuple
- measurementNames()¶
- Returns:
The names of the measurements.
- Return type:
list of type str
- metatext()¶
- Returns:
The metatext of the object or None if no metatext is set.
- Return type:
str | None
- nlinfo()¶
- Returns:
The trajectory information.
- Return type:
dict
- nlprint(stream=<_io.TextIOWrapper name='<stdout>' mode='w' encoding='utf-8'>)¶
Write a text description of the trajectory to the stream.
- Parameters:
stream (file-like object) – The stream to write to. Default:
sys.stdout
- numberOfAtoms()¶
- Returns:
The number of atoms in the system.
- Return type:
int
- potentialEnergies(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The potential energies of the images or None the trajectory length is zero.
- Return type:
PhysicalQuantity of type energy | None
- pressureTensors(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The pressure tensors of the images as a vector with shape (m, 3, 3) or None if the trajectory length is zero.
- Return type:
PhysicalQuantity of type pressure | None
- pressures(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The pressure values of the images as a vector with shape (m) or None if the trajectory length is zero.
- Return type:
PhysicalQuantity of type pressure | None
- prune(interval, path=None, object_id=None)¶
Copy this MDTrajectory and reduce the number of images.
- Parameters:
interval (int) – Only keep every interval steps.
path (str | None) – The path to the HDF5 file. If the path is None then all data will be stored in memory and no file will be written. Default: None
object_id – The object id to use. If the value is None then an id will be automatically made. Default: None
- Returns:
The pruned MDTrajectory.
- Return type:
- reservoirPressures(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The reservoir pressure of the MD barostat. If the trajectory length is zero or this trajectory was not generated from a NPT simulation then None is returned.
- Return type:
PhysicalQuantity of type temperature
- reservoirTemperatures(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The reservoir temperature of the MD thermostat. If the trajectory length is zero or this trajectory was generated from a NVE simulation then None is returned.
- Return type:
PhysicalQuantity of type temperature
- setMetatext(text)¶
Set a given metatext string on the object.
- Parameters:
text (str | None) – The metatext string that should be set. A value of “None” can be given to remove the current metatext.
- steps(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The MD step numbers of the images or None the trajectory length is zero.
- Return type:
array of type int | None
- stresses(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The stresses of the images. If the trajectory length is zero or the stresses were not written during the MolecularDynamics run then None is returned. None is also returned if the configuration is a MoleculeConfiguration.
- Return type:
PhysicalQuantity of type pressure | None
- temperatures(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The instantaneous kinetic temperatures of the images. If the trajectory length is zero or the temperatures were not written during the MolecularDynamics run then None is returned.
- Return type:
PhysicalQuantity of type temperature
- timeInterval()¶
- Returns:
The time interval between two images.
- Return type:
PhysicalQuantity of type time
- timeStep()¶
- Returns:
The MD time step of the simulation.
- Return type:
PhysicalQuantity of type time
- times(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The simulation times of the images or None the trajectory length is zero.
- Return type:
PhysicalQuantity of type time | None
- trajectoryInterval()¶
- Returns:
The interval at which snapshots are saved.
- Return type:
int
- uniqueElements()¶
- Returns:
The unique elements of the images or None if the trajectory length is zero.
- Return type:
list of type
PeriodicTableElement
- velocities(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: All indices
- Returns:
The velocities of the images. If the trajectory length is zero or the velocities were not written during the MolecularDynamics run then None is returned.
- Return type:
PhysicalQuantity of type length/time
- volumes(idx=Ellipsis)¶
- Parameters:
idx (int | slice) – The index or slice to return. Default: Ellipsis
- Returns:
The volumes of the unit cells. If this was a MD simulation of molecules, then None will be returned.
- Return type:
PhysicalQuantity of type length**3 | None
- writeToXYZ(filename)¶
Make a xyz dump of the whole trajectory.
- Parameters:
filename (str) – The name of the file to write to.
Usage Examples¶
Perform four Molecular Dynamics simulations with different time steps and save their trajectories in the file trajectory.nc.
# Define a benzene ring
cartesian_coordinates = [[ 0.0 , 1.399795, 0.0],
[ 1.212253, 0.69976 , 0.0],
[ 1.212253, -0.69976 , 0.0],
[ 0.0 , -1.399795, 0.0],
[-1.212253, -0.69976 , 0.0],
[-1.212253, 0.69976 , 0.0],
[ 0.0 , 2.500678, 0.0],
[ 2.165671, 1.250363, 0.0],
[ 2.165671, -1.250363, 0.0],
[ 0.0 , -2.500678, 0.0],
[-2.165671, -1.250363, 0.0],
[-2.165671, 1.250363, 0.0]]*Angstrom
molecule_configuration = MoleculeConfiguration(
elements=[Carbon,]*6+[Hydrogen,]*6,
cartesian_coordinates=cartesian_coordinates
)
# Attach a calculator
molecule_configuration.setCalculator(BrennerCalculator())
initial_velocity = MaxwellBoltzmannDistribution(
temperature=300.0*Kelvin
)
length_of_simulation = 20*femtoSecond
# Generate trajectories for different time steps
for time_step in [0.1, 0.5, 1, 2]*femtoSecond:
method = NVEDynamics(
time_step=time_step,
initial_velocity=initial_velocity
)
# Generate trajectory
molecular_dynamics = MolecularDynamics(
molecule_configuration,
trajectory_filename='trajectory.nc',
steps=int(length_of_simulation/time_step),
log_interval=max(1,int(2*femtoSecond/time_step)),
method=method
)
Plot the total energy as function of time for the trajectories in the file trajectory.nc.
import pylab
# Read in a list with the trajectories
trajectories = nlread('trajectory.nc', MDTrajectory)
pylab.figure()
for trajectory in trajectories:
# Get the time axis of the trajectory
t = trajectory.times().inUnitsOf(femtoSecond)
# Calculate the time step
time_step = t[-1]/trajectory.steps()[-1]
# Get the energies
epot = trajectory.potentialEnergies().inUnitsOf(eV)
ekin = trajectory.kineticEnergies().inUnitsOf(eV)
# Plot the data
pylab.plot(t,epot+ekin, label='timestep=' + str(time_step)+' fs')
pylab.xlabel('time (fs)')
pylab.ylabel('total energy (eV)')
pylab.legend()
pylab.show()
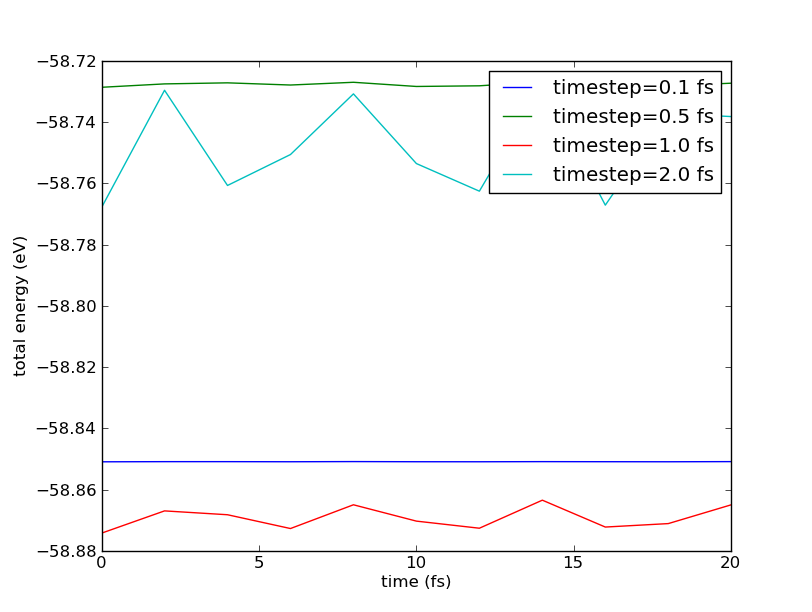
Fig. 188 The total energy as function of the simulation time in MD runs with different time steps.¶