PolymerSegmentAnalyzer¶
Included in QATK.Polymers
- class PolymerSegmentAnalyzer(md_trajectory, polymer_tags=None, end_tags=None, start_time=None, end_time=None, time_resolution=None, atoms_per_segment=None, gui_worker=None)¶
Create an anylzer to calculate the segment distances in polymer chains.
- Parameters:
md_trajectory (
MDTrajectory|AtomicConfiguration) – The MDTrajectory or AtomicConfiguration that contains the polymers.polymer_tags (list of type str | None | Automatic) – A list of tags for the polymers. If not specified default tags are used. Tags can also be set automatically with the flag Automatic
end_tags (list of type str | None) – A list of tags for the ends of each polymer chain. If not specified default tags are used.
start_time (PhysicalQuantity of the type time) – The start time. Default:
0.0 * fs.end_time (PhysicalQuantity of the type time) – The end time. Default: The last time image.
time_resolution (PhysicalQuantity of type time) – The time interval between snapshots in the MD trajectory that are included in the analysis.
atoms_per_segment (int) – The number of atoms that make up one monomer or segment. Default: 1.
gui_worker (PolymerAnalysisWorker) – Handle to the worker, for displaying the calculation progress. Default: None.
- averageSegmentLength(polymer_index)¶
Return the average segment length for the given polymer.
- Parameters:
polymer_index (int) – An index to the polymer chain, based on the order specified by the polymer chain tags.
- Returns:
The average segment length.
- Return type:
PhysicalQuantity of type length
- backboneAtomIndexes(polymer_index)¶
Return the atomic indexes of the backbone atoms for the given polymer.
- Parameters:
polymer_index (int) – An index to the polymer chain, based on the order specified by the polymer chain tags.
- Returns:
The index of the backbone atoms for the given polymer chain.
- Return type:
list of int
- bondCorrelation(polymer_index)¶
Return the bond correlation function for the segments for the given polymer.
- Parameters:
polymer_index (int) – An index to the polymer chain, based on the order specified by the polymer chain tags.
- Returns:
The bond correlation for the given polymer chain.
- Return type:
ndarray
- characteristicRatio(polymer_index)¶
Return the characteristic ratio for the given polymer chain.
- Parameters:
polymer_index (int) – An index to the polymer chain, based on the order specified by the polymer chain tags.
- Returns:
The characteristic array, index by segment distance, for the given polymer chain.
- Return type:
ndarray
- polymerTags()¶
Return the polymer tags used for the analysis.
- Returns:
A list of the polymer tags.
- Return type:
list
- rmsSegmentDistance(polymer_index)¶
Return the RMS distance between segments separated by a certain number of segments. These distances are averaged over the entire polymer chain
- Parameters:
polymer_index (int) – An index to the polymer chain, based on the order specified by the polymer chain tags.
- Returns:
The RMS segment-segment distance for the given polymer chain.
- Return type:
PhysicalQuantity of type length
- segmentPositions(polymer_index)¶
Return the positions of the segments for the given polymer.
- Parameters:
polymer_index (int) – An index to the polymer chain, based on the order specified by the polymer chain tags.
- Returns:
The index of the segment positions for the given polymer chain.
- Return type:
PhysicalQuantity of type length
- times()¶
- Returns:
Times for each image used in the analysis.
- Return type:
PhysicalQuantityof type time
Usage Examples¶
Calculate the bond correlation and characteristic ratio of a polystyrene melt during a molecular dynamics simulation starting from a non-equilibrated structure.
# -------------------------------------------------------------
# Initial Configuration
# -------------------------------------------------------------
bulk_configuration = nlread('Polystyrene.hdf5')[-1]
# -------------------------------------------------------------
# OPLS-AA Calculator
# -------------------------------------------------------------
potential_builder = OPLSPotentialBuilder()
calculator = potential_builder.createCalculator(bulk_configuration)
bulk_configuration.setCalculator(calculator)
# -------------------------------------------------------------
# Molecular Dynamics
# -------------------------------------------------------------
initial_velocity = ConfigurationVelocities(
remove_center_of_mass_momentum=True
)
method = NPTMartynaTobiasKlein(
time_step=1*femtoSecond,
reservoir_temperature=300*Kelvin,
reservoir_pressure=1*bar,
thermostat_timescale=100*femtoSecond,
barostat_timescale=500*femtoSecond,
initial_velocity=initial_velocity,
heating_rate=0*Kelvin/picoSecond,
)
constraints = [FixCenterOfMass()]
md_trajectory = MolecularDynamics(
bulk_configuration,
constraints=constraints,
trajectory_filename='Polystyrene_Trajectory.hdf5',
steps=100000,
log_interval=10,
trajectory_interval=1000,
method=method
)
# -------------------------------------------------------------
# Calculate End to End Distance
# -------------------------------------------------------------
analyzer = PolymerSegmentAnalyzer(
md_trajectory,
start_time=0*ps,
end_time=100*ps,
end_tags=['HEAD_CONNECT', 'TAIL_CONNECT'],
atoms_per_segment=2
)
number_of_segments = 64
number_of_chains = 8
bond_correlation = numpy.zeros(number_of_segments-1)
characteristic_ratio = numpy.zeros(number_of_segments)
segments = numpy.arange(number_of_segments)
for i in range(number_of_chains):
bond_correlation += analyzer.bondCorrelation(i)
characteristic_ratio += analyzer.characteristicRatio(i)
bond_correlation /= number_of_chains
characteristic_ratio /= number_of_chains
# -------------------------------------------------------------
# Plot The Results
# -------------------------------------------------------------
import pylab
pylab.figure()
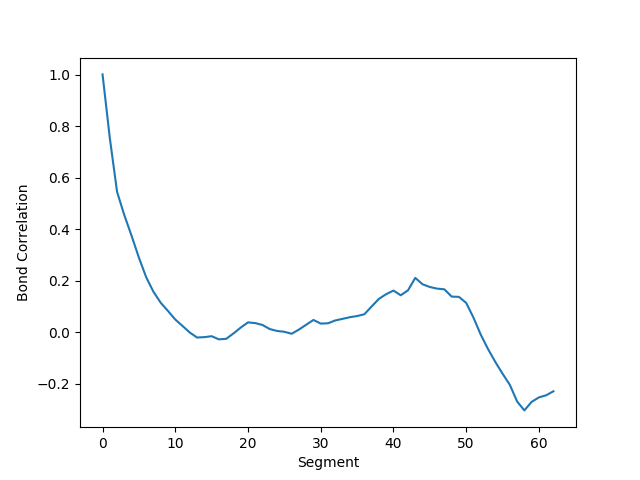
pylab.plot(segments[:number_of_segments-1], bond_correlation)
pylab.xlabel('Segment')
pylab.ylabel('Bond Correlation')
pylab.savefig('Bond_Correlation_Plot.png')
pylab.figure()
pylab.plot(segments, characteristic_ratio)
pylab.xlabel('Segment')
pylab.ylabel('Characteristic Ratio')
pylab.savefig('Characteristic_Ratio_Plot.png')
PolymerSegmentAnalyzer_Example.py
Polystyrene.hdf5
The script will run a short molecular dynamics calculation from the given starting structure and then calculate bond correlation function and the characteristic ratio for the polymer system. This is shown in the two plots below.

Notes¶
This class enables the calculation of the details of the arrangements of different polymer backbone
segments from a MDTrajectory or BulkConfiguration.
From a statistical point of view, a polymer can be viewed as a number of segments forming a chain. These segments may be atoms or monomer units. Ideally this chain should form a self-avoiding random walk, however the chemical nature of the polymer system causes deviations from this ideal case. Analyzing the relative positions of the polymer segments allows the deviations from ideal to be characterized. It can also give insight into the representative quality of the polymer structure[1].
Information about the polymer segments is calculated from the backbone atoms. These are defined as the atoms that lie on the bonded path between the two ends of the polymer chain. When segments are constructed from multiple atoms, the segment position is taken as the average position of the atoms. One important measure of the polymer chain is the bond-bond correlation, \(R_{bb}\), which gives a measure of the decay of geometrical information along the polymer chain. This can be given as:
Here \(\mathbf{b}_i\) is the ith inter-segment distance and \(\left<b\right>\) is the average inter-segment distance.
Another important property is the average square distance between segments separated by \(n\) bonds, \(\left<r^2_n\right>\). This property can be used to calculate the characteristic ratio \(C_n\), which can be given as[2]:
In well equilibrated chains the value of \(C_n\) should asymptote to the value \(C_\infty\) at large values of \(n\)
To calculate the polymer segment descriptors a PolymerSegmentAnalyzer object first needs
to be created. This takes a MDTrajectory or BulkConfiguration and polymer
segment properties for the specified configurations. The specific frames in the
MDTrajectory to analyzed can be selected using the start_time, end_time and
time_resolution arguments. The polymer chains analyzed in the configuration can be specified
with the polymer_tags argument. This takes a list of tags of polymer molecules in the
configuration. By default polymers tagged with POLYMER_MOLECULE_# are analyzed, where # is
a numerical index. Tags indicating the start and end of each polymer chain also need to be added.
The argument end_tags takes a list of strings that contain the tags for the end atoms. These
tags can be specified in any order. By default the tags HEAD_CONNECT and TAIL_CONNECT are
used. All the default polymer and end tags are placed on polymer configurations built with the
PolymerMonteCarloBuilder. It is also possible to control the size of each polymer
segment using the atoms_per_segment argument. Using this argument monomer units and be
considered as comprising segments, rather than individual atoms.
Once the PolymerSegmentAnalyzer object is created and the values of each of the
descriptors has been calculated, the values can be queried using the remaining functions. Each
function takes a required polymer_index argument, which specifies the index of the polymer chain
for which the data is sought. Data for each chain is returned separately to allow for systems with
chains of different lengths. The indices of the atoms that make up the backbone of each polymer
can be found using the backboneAtomIndexes() function. Likewise the
Cartesian positions of each segment is returned by the
segmentPositions() function. When segments consist of more than one
atom these positions will be distinct from the backbone atomic positions. The average segment
length of each polymer is returned by the averageSegmentLength()
function. These functions all return arrays of data, indexed first by the image in the molecular
dynamics trajectory that the data is calculated from.
Other available descriptors of the polymer system are averaged over the selected molecular dynamics
images, and so the data returned by these functions is indexed over the integer distance between
segments. One such function is the bondCorrelation() function, which
returns the bond correlation function for the polymer averaged over the available molecular dynamics
images. In a similar manner rmsSegmentDistance() returns the
square root of the average square distance, and characteristicRatio()
returns the previously defined characteristic ratio.
In cases where all of the polymers in the system are the same length, it is possible to easily average these properties over the polymer chains. The included example illustrates how this may be performed for the bond correlation function and the characteristic ratio.