HamiltonianDerivatives¶
Included in QATK.Analysis
- class HamiltonianDerivatives(configuration, filename, object_id, repetitions=None, atomic_displacement=None, constraints=None, use_equivalent_bulk=None, constrain_electrodes=None, log_filename_prefix=None, processes_per_displacement=None, use_wigner_seitz_scheme=None, auxiliary_basis_set=None, compensate_fermi_level_shift=None)¶
Constructor for the HamiltonianDerivatives object.
- Parameters:
configuration (
BulkConfiguration|DeviceConfiguration) – The configuration for which to calculate the Hamiltonian derivatives.filename (str) – The full or relative path to save the results to. See
nlsave().object_id (str) – The object id to use when saving. See
nlsave().repetitions (
Automatic| list of ints) – The number of repetitions of the system in the A, B, and C-directions given as a list of three positive integers, e.g.[3, 3, 3], orAutomatic. Each repetition value must be odd. Default:(1, 1, 1)atomic_displacement (PhysicalQuantity of type length) – The distance the atoms are displaced in the finite difference method. Default:
0.01 * Angstromconstraints (list of type int) – List of atomic indices that will be constrained, e.g.
[0, 2, 10]. Default: Empty list[]use_equivalent_bulk (bool) – Control if a
DeviceConfigurationshould be treated as aBulkConfiguration. Default:Trueconstrain_electrodes (bool) – Control if the electrodes and electrode extensions should be constrained in case of a
DeviceConfiguration. Default:Falselog_filename_prefix (str or None) – Prefix for the filenames where the logging output for every displacement calculation is stored. The filenames are formed by appending a number and the file extension (“.log”). Default:
"hamiltonian_displacement_"processes_per_displacement (int |
ProcessesPerNode) – The number of processes assigned to calculating a single displacement. Default:ProcessesPerNodeuse_wigner_seitz_scheme (bool) – Control if the real space HamiltonianDerivative Matrix should be extended according to the Wigner Seitz construction.
use_wigner_seitz_scheme=Trueis only supported for simple orthorhombic, simple tetragonal and simple cubic lattices. Default:Falseauxiliary_basis_set (list | tuple |
BasisSet) – A different (typically smaller) basis set to be used for evaluating the derivative of the effective potential. This auxiliary basis set will be used in the self-consistent updates, and can potentially speed up the calculations significantly, although at the cost of accuracy. The Hamiltonian derivatives matrices will be evaluated in the basis set on the calculator. This option is only available for LCAOCalculators. Default:Nonei.e. no auxiliary basis set is used.compensate_fermi_level_shift (bool) –
If
True, possible changes in the Fermi level due to finite size effects are compensated, according to eq.17 in ref. [1]. This requires the evaluation of the Fermi level for each displaced configuration. In the case of non-selfconsistent semi-empirical calculations, setting it toFalsewill give a large speedup.Note: for
LCAOCalculatorthis parameter is ignored, as the hamiltonian derivatives are calculated differently, and it is not necessary to adjust for finite size effects. Default:True
- atomicDisplacement()¶
- Returns:
The distance the atoms are displaced in the finite difference method.
- Return type:
PhysicalQuantity with length unit.
- auxiliaryBasisSet()¶
- Returns:
A different (typically smaller) basis set to be used for evaluating the derivative of the effective potential.
- Return type:
list | tuple |
BasisSet
- compensateFermiLevelShift()¶
- Returns:
Whether a change in Fermi level for displaced configurations should be compensated.
- Return type:
bool
- constrainElectrodes()¶
- Returns:
Boolean determining if the electrodes and electrode extensions are constrained in case of a DeviceConfiguration.
- Return type:
bool
- constraints()¶
- Returns:
The list of constrained atoms.
- Return type:
list of int
- dependentStudies()¶
- Returns:
The list of dependent studies.
- Return type:
list of
Study
- filename()¶
- Returns:
The filename where the study object is stored.
- Return type:
str
- logFilenamePrefix()¶
- Returns:
The filename prefix for the logging output of the study.
- Return type:
str |
LogToStdOut
- nlinfo()¶
- Returns:
Structured information about the HamiltonianDerivatives.
- Return type:
dict
- nlprint(stream=None)¶
Print a string containing an ASCII table useful for plotting the Study object.
- Parameters:
stream (python stream) – The stream the table should be written to. Default:
NLPrintLogger()
- numberOfProcessesPerTask()¶
- Returns:
The number of processes to be used to execute each task. If None, all available processes execute each task collaboratively.
- Return type:
int | None |
ProcessesPerNode
- numberOfProcessesPerTaskResolved()¶
- Returns:
The number of processes to be used to execute each task. Default values are resolved based on the current execution settings.
- Return type:
int
- objectId()¶
- Returns:
The name of the study object in the file.
- Return type:
str
- processesPerDisplacement()¶
- Returns:
The number of processes per displacement.
- Return type:
int |
ProcessesPerNode
- repetitions()¶
- Returns:
The number of repetitions of the system in the A, B, and C-directions.
- Return type:
list of three int.
- saveToFileAfterUpdate()¶
- Returns:
Whether the study is automatically saved after it is updated.
- Return type:
bool
- uniqueString()¶
Return a unique string representing the state of the object.
- update()¶
Run the calculations for the HamiltonianDerivatives study object.
- useEquivalentBulk()¶
- Returns:
Boolean determining if a DeviceConfiguration is treated as a BulkConfiguration.
- Return type:
bool.
- wignerSeitzScheme()¶
- Returns:
Boolean to control if the real space Dynamical Matrix should be extended according to the Wigner-Seitz construction.
- Return type:
bool
Usage Examples¶
Note
Study objects behave differently from analysis objects. See the Study object overview for more details.
Calculate the Hamiltonian derivatives for a system repeated five times in the B direction and three times in the C direction.
hamiltonian_derivatives = HamiltonianDerivatives(
configuration,
filename='HamiltonianDerivatives.hdf5',
object_id='hamiltonian_derivatives',
repetitions=(1,5,3),
)
hamiltonian_derivatives.update()
When using repetitions=Automatic, the cell is repeated such that all atoms
within a pre-defined, element-pair dependent interaction range are included.
hamiltonian_derivatives = HamiltonianDerivatives(
configuration,
filename='HamiltonianDerivatives.hdf5',
object_id='hamiltonian_derivatives',
repetitions=Automatic,
)
hamiltonian_derivatives.update()
The default number of repetitions i.e. repetitions=Automatic can be found
before a calculation using the function checkNumberOfRepetitions().
(nA, nB, nC) = checkNumberOfRepetitions(configuration)
Notes¶
The Hamiltonian derivatives are calculated using the central finite difference method in a repeated cell constituting a super cell. That is, the Hamiltonian derivatives are calculated for each atom in the central cell by displacing the atom in the supercell and determining the Hamiltonian for two displacements from its original position along each of the Cartesian directions. In DFT, the derivatives of the Hamiltonian \(\hat{H}\) can be expressed as the derivatives of the effective potential \(V_{\text{eff}}\) since the kinetic term does not contribute. Thus the Hamiltonian derivatives for the \(i\) and \(j\) basis functions can be approximated as
where \(R_{I, \alpha}\) is the \(\alpha\) cartesian coordinate for atom \(I\) in the central unit cell, and \(\delta\) is the atomic displacement. For Slater-Koster calculators, the Hamiltonian derivatives are described by the on-site and off-site parameters and their distance dependence, hence
Terminated HamiltonianDerivatives calculations can be resumed by re-running the
same script or reading the study object from file and calling update() on
it. The study object will automatically detect which displacement calculations
have already been carried out and only run the calculations that are not yet
completed.
Notes for DFT¶
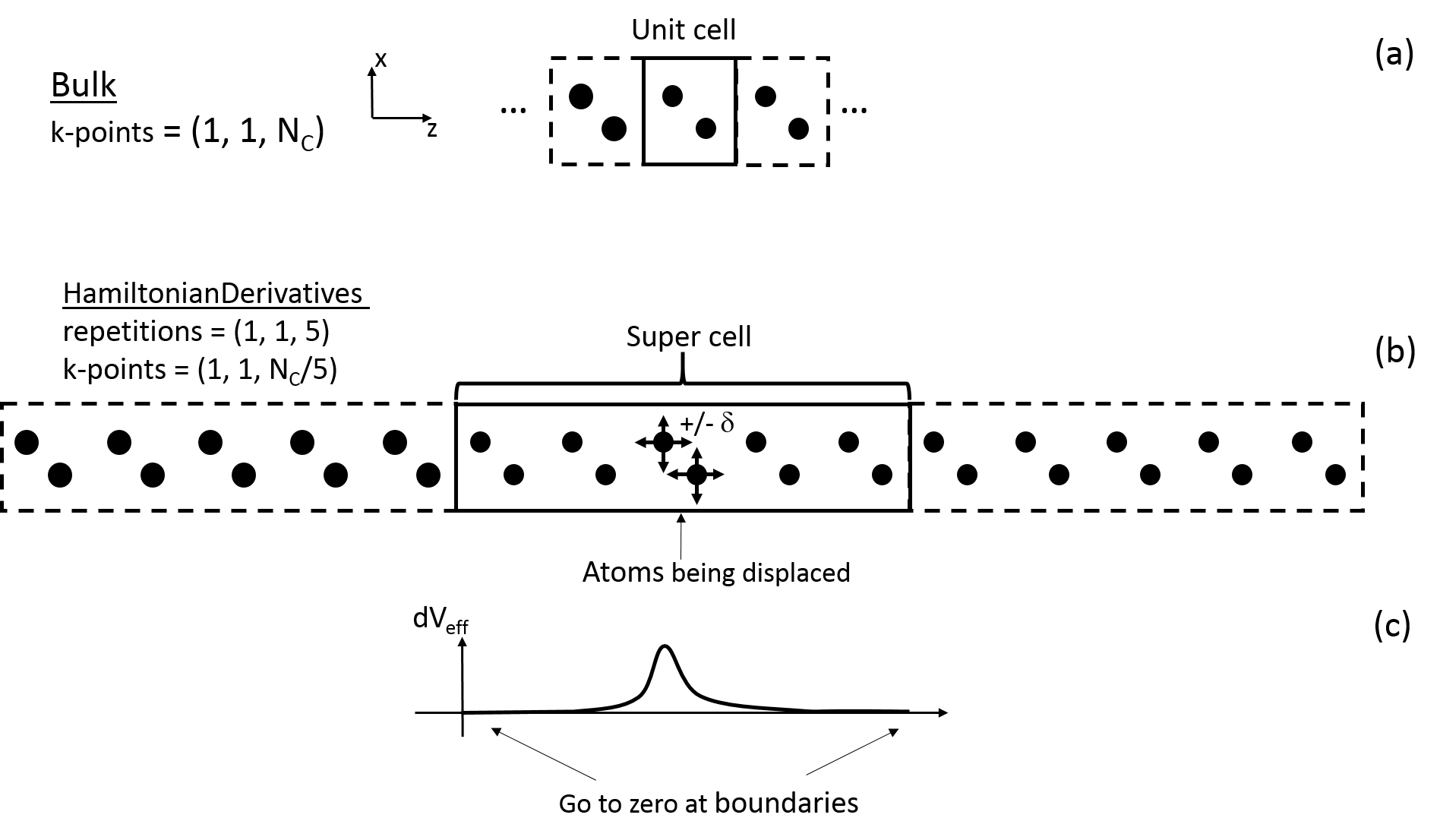
Fig. 179 (a) a unit cell for a 1D system confined in the \(x\) and \(y\) -directions. The k-point sampling is correspondingly \((1, 1, N_{\text{C}})\) , where \(N_{\text{C}}\) is the sampling in the \(z\) -direction. (b) a super cell created by five repetitions in the \(z\) -direction of the unit cell in (a). In the calculation of the Hamiltonian derivatives the atoms in the center unit cell of the super cell (the two atom indicated with arrows) are displaced by \(\pm\delta\) in each of the Cartesian directions. (c) the change in the effective potential \(\text{d} V_{\text{eff}}\) in the super cell as an atom is displaced \(\delta\) . The number of repetitions must ensure that the change in the effective potential goes to zero at the boundaries of the super cell in the non-confined directions for every atomic displacement.¶
When calculating the Hamiltonian derivatives both the number of sampled k-points for the super cell and repetitions in the confined the directions must be 1. In the following the number of sampled k-points for the super cell and repetitions in the non-confined directions will be adressed. To simplify things a system with only one non-confined direction will be used, see Fig. 179 (a), but the relations for the one non-confined direction applies to all non-confined directions. The bulk configuration in Fig. 179 (a) is converged in the total energy with respect to the number of k-points of \((1, 1, N_{\text{C}})\). The number of repetitions in the super cell in the non-confined direction, see Fig. 179 (b), is chosen large enough such that the change in the effective potential \(\text{d} V_{\text{eff}}\) goes to zero at the boundaries of the super cell in the non-confined directions for every atomic displacement, confer Fig. 179 (c). For this system five repetitions of the unit cell in the confined direction is enough for the change in the effective potential to go to zero at the boundaries. The recommended k-point sampling for the non-confined direction is the number of k-points in the non-confined direction for the unit cell divided by the number of repetitions in the non-confined direction. The k-point sampling then becomes \((1, 1, \frac{N_{\text{C}}}{\text{repetitions in C}})\) and in this particular case \((1, 1, N_{\text{C}}/5)\) .
Note
From QuantumATK-2019.03 onwards, the k-point sampling and
density-mesh-cutoff will be automatically adapted to the given number
of repetitions when setting up the super cell inside
DynamicalMatrix and HamiltonianDerivatives. That
means you can specify the calculator settings for the unit cell and use it
with any desired number of repetitions in dynamical matrix and hamiltonian
derivatives calculations.
The Hamiltonian derivatives calculations generally requires a low tolerance in the
IterationControlParameters settings, e.g. a tolerance of 1e-6. Finally,
it should be noted that the HuckelCalculator currently does not support
calculation of the Hamiltonian derivatives.