MolecularOrderParameter¶
Included in QATK.Polymers
- class MolecularOrderParameter(md_trajectory, polymer_tags=None, start_time=None, end_time=None, time_resolution=None, gui_worker=None)¶
Analyzer for the rotational and translational order in a molecular structure.
- Parameters:
md_trajectory (
MDTrajectory|MoleculeConfiguration|BulkConfiguration|DeviceConfiguration|SurfaceConfiguration) – The MDTrajectory or AtomicConfiguration that contains the polymers.polymer_tags (list of type str | None | Automatic) – A list of tags for the polymers. If not specified default tags are used. Tags can also be set automatically with the flag Automatic
start_time (PhysicalQuantity of the type time) – The start time. Default:
0.0 * fs.end_time (PhysicalQuantity of the type time) – The end time. Default: The last time image.
time_resolution (PhysicalQuantity of type time) – The time interval between snapshots in the MD trajectory that are included in the analysis.
gui_worker (PolymerAnalysisWorker) – Handle to the worker, for displaying the calculation progress. Default: None.
- polymerTags()¶
Return the polymer tags used for the analysis.
- Returns:
A list of the polymer tags.
- Return type:
list
- rotationalOrder()¶
- Returns:
The rotational order parameter for each image in the given trajectory.
- Return type:
ndarray
- times()¶
- Returns:
Times for each image used in the analysis.
- Return type:
PhysicalQuantityof type time
- translationalOrder()¶
- Returns:
The translational order parameter for each image in the given trajectory.
- Return type:
ndarray
Usage Examples¶
Calculate the translational and rotational order of a polystyrene melt during a molecular dynamics simulation starting from a non-equilibrated structure.
from NL.GUI.Graphics.Plotter.API import *
# -------------------------------------------------------------
# Initial Configuration
# -------------------------------------------------------------
bulk_configuration = nlread('Polystyrene.hdf5')[-1]
# -------------------------------------------------------------
# OPLS-AA Calculator
# -------------------------------------------------------------
potential_builder = OPLSPotentialBuilder()
calculator = potential_builder.createCalculator(bulk_configuration)
bulk_configuration.setCalculator(calculator)
# -------------------------------------------------------------
# Molecular Dynamics
# -------------------------------------------------------------
initial_velocity = ConfigurationVelocities(
remove_center_of_mass_momentum=True
)
method = NPTMartynaTobiasKlein(
time_step=1*femtoSecond,
reservoir_temperature=300*Kelvin,
reservoir_pressure=1*bar,
thermostat_timescale=100*femtoSecond,
barostat_timescale=500*femtoSecond,
initial_velocity=initial_velocity,
heating_rate=0*Kelvin/picoSecond,
)
constraints = [FixCenterOfMass()]
md_trajectory = MolecularDynamics(
bulk_configuration,
constraints=constraints,
trajectory_filename='Polystyrene_Trajectory.hdf5',
steps=100000,
log_interval=10,
trajectory_interval=1000,
method=method
)
# -------------------------------------------------------------
# Calculate Order Parameters
# -------------------------------------------------------------
analyzer = MolecularOrderParameter(md_trajectory)
# -------------------------------------------------------------
# Plot The Results
# -------------------------------------------------------------
model = PlotModel(ps)
model.axis('x').setLabel('Time')
model.axis('y').setLabel('Order Parameter')
model.legend().setVisible(True)
colors = ['red', 'blue']
line = Line(analyzer.times(), analyzer.rotationalOrder())
line.setLabel('Rotation')
line.setColor('red')
model.addItem(line)
line = Line(analyzer.times(), analyzer.translationalOrder())
line.setLabel('Translation')
line.setColor('blue')
model.addItem(line)
model.setLimits('x', 0*ps, 100*ps)
model.setLimits('y', 0, 1)
save(model, 'MolecularOrderParameter_Plot.png')
MolecularOrderParameter_Example.py
Polystyrene.hdf5
The script will run a short molecular dynamics calculation from the given starting structure and then calculate the molecular order for the polymer system. The resulting order is shown in the plot below.
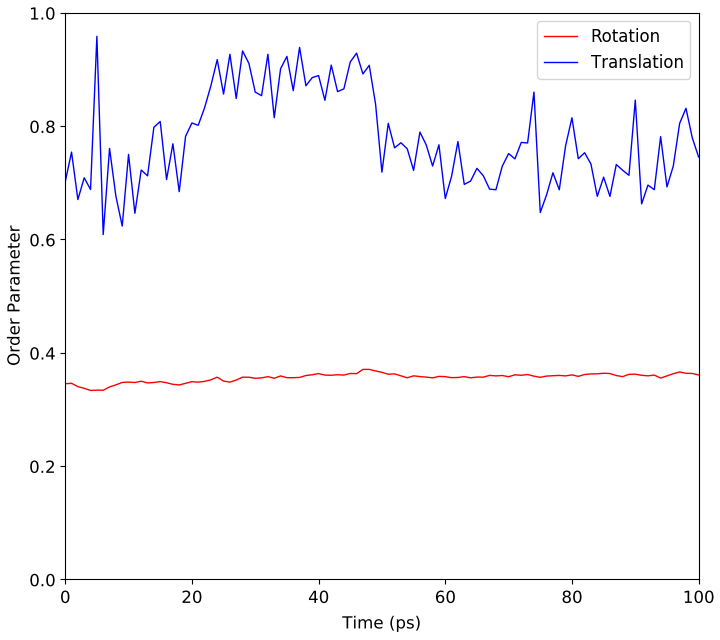
Fig. 189 Molecular rotational and translational parameters for polystyrene structure.¶
Notes¶
This class enables the calculation of the relative rotational and translational order of either
molecules or polymer chains from a MDTrajectory or BulkConfiguration.
Molecules in a bulk system can be both translationally or rotationally ordered. Translational order relates to the ability for molecules to form regularly spaced layers along a particular direction. Likewise rotational order relates to the extent that molecules in the bulk structure are aligned together[1]. To calculate the ordering of a molecular system, the vector representing each molecule is first found by diagonalizing the inertial tensor \(\mathbf{I}\), which is defined as
Here \(\mathbf{r}_j\) is the distance from the molecule center of mass to the jth atom and \(m_j\) is the mass of that atom. The long axis of the molecule that defines its direction is the eigenvector associated with the smallest eigenvalue. With the direction of each molecule defined, an overall ordering direction can be found similarly by diagonalizing the second-rank ordering tensor \(\mathbf{Q}\), which is given as:
Here \(\mathbf{u}_j\) is a unit vector representing the direction of the jth molecule. The nematic direction is then the eigenvector associated with the largest eigenvalue of the ordering tensor. Given the unit vectors \(\mathbf{u}_j\) and \(\mathbf{n}\) representing the direction of each molecule and the overall ordering direction respectively, the rotational ordering \(\omega\) is given as:
The translational ordering \(\tau\) can also be given as
Here \(d\) represents the interlayer spacing. The interlayer spacing is found by determining the spacing that maximizes the translational order.
Both rotational and translational ordering parameters lie in the range 0 to 1 for completely disordered materials and fully ordered materials, respectively.
To calculate the molecular order a MolecularOrderParameter object first needs
to be created. This takes a MDTrajectory or BulkConfiguration. The specific
images in the MDTrajectory to be analyzed can be selected using the start_time,
end_time and time_resolution arguments. The molecules analyzed in the configuration can be
specified with the polymer_tags argument. This takes a list of tags of polymer molecules in the
configuration. By default polymers tagged with POLYMER_MOLECULE_# are analyzed, where # is
a numerical index. For molecular systems it is also possible to give the flag Automatic for the
polymer_tags argument. This specifies that the molecular bond graph, rather than explicit tags,
are to be used to determine which atoms belong to each molecule.
Once the MolecularOrderParmeter object is created and the values of each of the
descriptors has been calculated, the values can be queried using the remaining functions. The
method rotationalOrder returns the numpy array of the rotational order parameters for each
image in the trajectory. Likewise the translationalOrder method returns a numpy array with the
translational ordering values.