InelasticTransmissionSpectrum¶
Included in QATK.Analysis
- class InelasticTransmissionSpectrum(configuration, dynamical_matrix=None, hamiltonian_derivatives=None, bulk_dynamical_matrix=None, bulk_hamiltonian_derivatives=None, energies=None, kpoints=None, qpoints=None, method=None, self_energy_calculator=None, energy_zero_parameter=None, infinitesimal=None, phonon_modes=None, phonon_energy_minimum=None, phonon_energy_maximum=None, phonon_energy_intervals=None, fermis_golden_rule_only=None, spectral_representation=None, electrode_extensions=None, store_coupling_matrices=None, reuse_tolerance=None, voltage_fraction_left=None)¶
Class for calculating the inelastic transmission spectrum of a device configuration using the eXtended Lowest Order Expanssion (XLOE) method or Lowest Order Expanssion (LOE) method.
- Parameters:
configuration (
DeviceConfiguration) – The DeviceConfiguration for which the inelastic transmission should be calculated.dynamical_matrix (
DynamicalMatrix) – A DynamicalMatrix object constructed for the same DeviceConfiguration as this object. This option is mutually exclusive tobulk_dynamical_matrix.hamiltonian_derivatives (
HamiltonianDerivatives) – A HamiltonianDerivatives object constructed for the same DeviceConfiguration and calculator as this object. This option is mutually exclusive tobulk_hamiltonian_derivatives.bulk_dynamical_matrix (
DynamicalMatrix) – A DynamicalMatrix object constructed for the same DeviceConfiguration as this object. This option can only be used if the central region of the device configurationconfigurationcan be obtained asNrepetitions of the bulk configuration used to calculate thebulk_dynamical_matrix. This option is mutually exclusive todynamical_matrix.bulk_hamiltonian_derivatives (
HamiltonianDerivatives) – A HamiltonianDerivatives object constructed for a bulk configuration. This option can only be used if the central region of the device configurationconfigurationcan be obtained asNrepetitions of the bulk configuration used to calculate thebulk_hamiltonian_derivatives. This option is mutually exclusive tohamiltonian_derivatives.energies (A PhysicalQuantity list with an energy unit.) – The energy for which the inelastic transmission function is calculated. The energy is calculated relative to the Fermi energy. Default:
[0.0]*eVkpoints (sequence (size 3) of int |
MonkhorstPackGrid|KpointDensity|RegularKpointGrid| sequence of sequence (size 3) of float) – The k-points for the incoming electrons. This can either be given as a k-point grid or a list of fractional k-points, e.g.,[[0.0, 0.0, 0.0], [0.0, 0.0, 0.1], ...]. Default:[[0.0, 0.0, 0.0]]qpoints – The transverse q-points for which the phonons should be calculated. This can either be given as a q-point grid or a list of fractional q-points, e.g.,
[[0.0, 0.0, 0.0], [0.0, 0.0, 0.1], ...]. Outgoing electrons are scatered to k + q. Default:[[0.0, 0.0, 0.0]]method (
XLOE|LOE) – The method used for calculating the inelastic transmissions. Default:XLOEself_energy_calculator (
RecursionSelfEnergy|SparseRecursionSelfEnergy) – The self energy calculator to be used. Default:RecursionSelfEnergy(storage_strategy=NoStorage())energy_zero_parameter (
AverageFermiLevel|AbsoluteEnergy) – Specifies the choice for the energy zero. Default:AverageFermiLevelinfinitesimal (PhysicalQuantity with energy unit.) – Small energy, used to move the self-energy calculation away from the real axis. This is only relevant for recursion-style self-energy calculators. Default:
1.0e-6*eVphonon_modes (A list of non-negative integers.) – List with indices of phonon modes to consider. Default:
range(3*number_of_atoms). Include all modes.phonon_energy_minimum (PhysicalQuantity with energy unit.) – A lower limit for phonons to be considered. Default:
0.0*eVphonon_energy_maximum (PhysicalQuantity with energy unit.) – An upper limit for phonons to be considered. This could be the bias voltage, if “configuration” is calculated at finite bias voltages. Default:
1.0*eVphonon_energy_intervals (A PhysicalQuantity list with an energy unit.) – A list of energies. In each interval, between energy points i and i+1, all phonon modes will be summed to an effective phonon mode with an average energy of all the modes. Specifying this variable will overwrite the actual phonon modes. If the phonon_energy_intervals list has N energy points, there will effectively be N-1 phonon modes in the calculation for each (k,q) point. Default:
Nonefermis_golden_rule_only (bool) – If True, only the terms \(Tr(M A_L M A_R)\) are included in the inelastic transmissions. This is well-suited for e.g. p-i-n like structures. If true, then spectral_representation must be set to True (default). Default:
Falsespectral_representation (bool) – If True, a spectral representation will be used. Calculations will typically be much faster. Default:
Trueelectrode_extensions (list of two non-negative integers larger then 1 or [0,0]) – The left and right electrode will be extended by an integer number of full electrode sizes into the central region. The electrode extensions are treated as non-interacting. The electrode extensions must be short enough such that there is no direct overlap between the left and right electrode extensions. Default:
[0,0]store_coupling_matrices – Boolean to control if the electron-phonon coupling matrices should be precalculated and stored (True) or calculated on the fly (False). The option False saves memory, but is computationally slower. Default:
Truevoltage_fraction_left (float in range [0;1] with no unit.) – Voltage drop asymmetry used in XLOE. Voltage fraction at left electrode used in the energy shift in the Green’s functions \(G(E \pm \hbar\omega \cdot f_L)\) where \(f_L\) is the
voltage_fraction_left. 0.5 corresponds to a symmetric energy shift, while 1.0 means that all the energy shift is in the left chemical potential and 0.0 means that only the right electrode potential is shifted. Default:0.5
- elasticConductance(bias=None, temperature=None, fermi_shift=None, k_indices=None, spin=None)¶
Returns the elastic contribution to the differential conductance.
- Parameters:
bias (PhysicalQuantity of type voltage.) – The voltages for which the conductance should be calculated. Default:
numpy.linspace(0, 0.1, 100) * Volttemperature (PhysicalQuantity of type temperature.) – The temperature for which the conductance should be calculated. Default:
300 * Kelvinfermi_shift (PhysicalQuantity of type energy.) – Shift of Fermi level, such that the conductance is evaluated at the energy E = fermi_energy + fermi_shift. Default:
0.0 * eVk_indices (list.) – A list of non-negative integers with the indices for the k-points to include. Default: All the k-points, i.e. [0, 1, 2, …, number_of_kpoints-1]
spin (
Spin.Up|Spin.Down|Spin.All) – The spin flag. Default:Spin.Up
- Returns:
The elastic conductance for each bias point.
- Return type:
PhysicalQuantity of type Siemens.
- elasticCurrent(bias=None, temperature=None, fermi_shift=None, k_indices=None, spin=None)¶
Returns the elastic contribution to the current
- Parameters:
bias (PhysicalQuantity of type voltage.) – The voltages for which the current should be calculated. Default:
numpy.linspace(0,0.1,100)*Volttemperature (PhysicalQuantity of type temperature.) – The temperature for which the current should be calculated. Default:
300*Kelvinfermi_shift (PhysicalQuantity of type energy.) – Shift of Fermi level, such that the current is evaluated at the energy E = fermi_energy + fermi_shift. Default:
0.0*eVk_indices (list.) – A list of non-negative integers with the indices for the k-points to include. Default: All the k-points, i.e. [0, 1, 2, …, number_of_kpoints-1]
spin (
Spin.Up|Spin.Down|Spin.All) – The spin flag. Default:Spin.Up
- Returns:
The elastic current for each bias point.
- Return type:
PhysicalQuantity of type Ampere.
- elasticTransmission()¶
- Returns:
The elastic transmission of shape (N, K, S) where N is the number of energies, K is the number of k-points, and S is the number of spins.
- Return type:
numpy.ndarray.
- electrodeFermiLevels()¶
- Returns:
The electrode Fermi levels in absolute energies.
- Return type:
PhysicalQuantity of type energy.
- energies()¶
- Returns:
The energies at which the inelastic transmission spectrum is calculated.
- Return type:
PhysicalQuantity of energy unit.
- energyZero()¶
- Returns:
The energy zero used for the energy scale in this inelastic transmission spectrum.
- Return type:
PhysicalQuantity of type energy.
- energyZeroParameter()¶
- Returns:
The energy zero parameter used for the energy scale in this inelastic transmission spectrum.
- Return type:
AbsoluteEnergy|AverageFermiLevel
- evaluate()¶
- Returns:
Return a tuple with (energies, T0, Tsym_pos, Tsy,_neg, Tasym_pos, Tasym_neg), where
energies : is the list of energies given in the constructor
T0 : is the elastic transmission spectrum
Tsym_pos : is the symmetric contribution to the inelastic transmission at positive bias
Tsym_neg : is the symmetric contribution to the inelastic transmission at negative bias
Tasym_pos : is the asymmetric contribution to the inelastic transmission at positive bias
Tasym_neg : is the asymmetric contribution to the inelastic transmission at negative bias
- Return type:
tuple
- inelasticConductance(bias=None, temperature=None, modes=None, fermi_shift=None, k_indices=None, q_indices=None, spin=None, tolerance=None)¶
Returns the inelastic contribution to the differential conductance.
- Parameters:
bias (PhysicalQuantity of type voltage.) – The voltages for which the conductance should be calculated. Default:
numpy.linspace(0, 0.1, 100) * Volttemperature (PhysicalQuantity of type temperature.) – The temperature for which the conductance should be calculated. Default:
300 * Kelvinmodes (A list of non-negative integers.) – If specified, the inelastic conductance is only calculated for these modes. Default: All the modes, i.e. [0, 1, 2, …, N-1], where N is the number of degrees of freedom.
fermi_shift (PhysicalQuantity of type energy.) – Shift of Fermi level, such that the conductance is evaluated at the energy E = fermi_energy + fermi_shift. Default:
0.0 * eVk_indices (list.) – A list of non-negative integers with the indices for the k-points to include. Default: All the k-points, i.e. [0, 1, 2, …, number_of_kpoints-1]
q_indices (list.) – A list of non-negative integers with the indices for the q-points to include. Default: All the q-points, i.e. [0, 1, 2, …, number_of_qpoints-1]
spin (
Spin.Up|Spin.Down) – The spin flag. Default:Spin.Uptolerance (float.) – If specified, only phonon modes with an inelastic transmission larger than “tolerance*max(inelasticTransmission)” will be included. Default: 0.0 (all modes will be included).
- Returns:
The inelastic conductance for each bias point.
- Return type:
PhysicalQuantity of type Siemens.
- inelasticCurrent(bias=None, temperature=None, modes=None, fermi_shift=None, k_indices=None, q_indices=None, spin=None, tolerance=None)¶
Returns the inelastic contribution to the current.
- Parameters:
bias (PhysicalQuantity with voltage unit.) – The voltages for which the current should be calculated. Default:
numpy.linspace(0,0.1,100)*Volttemperature (PhysicalQuantity with temperature unit.) – The temperature for which the current should be calculated. Default:
300*Kelvinmodes (A list of non-negative integers.) – If specified, the inelastic current is only calculated for these modes. Default: All the modes, i.e. [0,1,2,…,N-1], where N is the number of degrees of freedom.
fermi_shift (PhysicalQuantity with energy unit.) – Shift of Fermi level, such that the current is evaluated at the energy E = fermi_energy + fermi_shift. Default:
0.0*eVk_indices (list.) – A list of non-negative integers with the indices for the k-points to include. Default: All the k-points, i.e. [0,1,2,…,number_of_kpoints-1]
q_indices (list.) – A list of non-negative integers with the indices for the q-points to include. Default: All the q-points, i.e. [0,1,2,…,number_of_qpoints-1]
spin (
Spin.Up|Spin.Down|Spin.All) – The spin flag. Default:Spin.Uptolerance (float.) – If specified, only phonon modes with an inelastic transmission larger than “tolerance*max(inelasticTransmission)” will be included. Default: 0.0 (all modes will be included).
- Returns:
The inelastic current for each bias point.
- Return type:
PhysicalQuantity of type Ampere.
- inelasticCurrentIntegral(bias=None, temperature=None, modes=None, fermi_shift=None, k_indices=None, q_indices=None, spin=None, tolerance=None, interpolation_factor=None)¶
Returns the inelastic contribution to the current calculate by integrating symmetric transmissions using the current expression from J. Appl. Phys. 109, 124503 (2011).
- Parameters:
bias (PhysicalQuantity of type voltage.) – The voltages for which the current should be calculated. Default:
numpy.linspace(0, 0.1, 100)*Volttemperature (PhysicalQuantity of type temperature.) – The temperature for which the transmission spectrum should be calculated. Default:
300 * Kelvinmodes (A list of non-negative integers.) – If specified, the inelastic current is only calculated for these modes. Default: All the modes, i.e. [0, 1, 2, …, N-1], where N is the number of degrees of freedom.
fermi_shift (PhysicalQuantity of type energy.) – Shift of Fermi level, such that the current is evaluated at the energy E = fermi_energy + fermi_shift. Default:
0.0 * eVk_indices (list.) – A list of non-negative integers with the indices for the k-points to include. Default: All the k-points, i.e. [0, 1, 2, …, number_of_kpoints-1]
q_indices (list.) – A list of non-negative integers with the indices for the q-points to include. Default: All the q-points, i.e. [0, 1, 2, …, number_of_qpoints-1]
spin (Spin.Up | Spin.Down |
Spin.All) – The spin flag. Default:Spin.Uptolerance (float.) – If specified, only phonon modes with an inelastic transmission larger than “tolerance*max(inelasticTransmission)” will be included. Default: 0.0 (all modes will be included).
interpolation_factor (int) – Integer determining interpolation refinement of the energies and transmissions. Default: 1
- Returns:
The inelastic current for each bias point.
- Return type:
PhysicalQuantity of type Ampere.
- inelasticElectronTunnelingSpectroscopy(bias=None, temperature=None, modes=None, fermi_shift=None, k_indices=None, q_indices=None, spin=None, tolerance=None)¶
Returns the inelastic tunneling spectrocopy signal (IETS), \(d^2I/dV^2\)
- Parameters:
bias (PhysicalQuantity of type voltage.) – The voltages for which the IETS should be calculated. Default:
numpy.linspace(0, 0.1, 100) * Volttemperature (PhysicalQuantity of type temperature.) – The temperature for which the IETS should be calculated. Default:
300 * Kelvinmodes (A list of non-negative integers.) – If specified, the IETS is only calculated for these modes. Default: All the modes, i.e. [0, 1, 2, …, N-1], where N is the number of degrees of freedom.
fermi_shift (PhysicalQuantity with energy unit.) – Shift of Fermi level, such that the IETS is evaluated at the energy E = fermi_energy + fermi_shift. Default:
0.0 * eVk_indices (list.) – A list of non-negative integers with the indices for the k-points to include. Default: All the k-points, i.e. [0, 1, 2, …, number_of_kpoints-1]
q_indices (list.) – A list of non-negative integers with the indices for the q-points to include. Default: All the q-points, i.e. [0, 1, 2, …, number_of_qpoints-1]
spin (
Spin.Up|Spin.Down|Spin.All) – The spin flag. Default:Spin.Uptolerance (float.) – If specified, only phonon modes with an inelastic transmission larger than “tolerance*max(inelasticTransmission)” will be included. Default: 0.0 (all modes will be included).
- Returns:
The inelastic tunneling spectrocopy signal for each bias point.
- Return type:
PhysicalQuantity of type Siemens/Volt.
- inelasticTransmissionAsymmetricNegativeBias(mode=None)¶
Return the asymmetric inelastic transmission for each mode for negative bias voltages.
- Parameters:
mode (int |
All) – The phonon mode for which to return the inelastic transmission. If not provided, the result for all modes will be returned. Default:All- Returns:
The asymmetric negative inelastic transmission. If mode=All, the shape of the returned array is (number_of_degrees_of_freedom, number_of_energies, number_of_kpoints, number_of_qpoints, number_of_spins). If mode=integer, the shape is (number_of_energies, number_of_kpoints, number_of_qpoints, number_of_spins).
- Return type:
numpy.ndarray.
- inelasticTransmissionAsymmetricPositiveBias(mode=None)¶
Return the asymmetric inelastic transmission for each mode for positive bias voltages.
- Parameters:
mode (int |
All) – The phonon mode for which to return the inelastic transmission. If not provided, the result for all modes will be returned. Default:All- Returns:
The asymmetric positive inelastic transmission. If mode=All, the shape of the returned array is (number_of_degrees_of_freedom, number_of_energies, number_of_kpoints, number_of_qpoints, number_of_spins). If mode=integer, the shape is (number_of_energies, number_of_kpoints, number_of_qpoints, number_of_spins).
- Return type:
numpy.ndarray.
- inelasticTransmissionSymmetricNegativeBias(mode=None)¶
Return the symmetric inelastic transmission for each mode for negative bias voltages.
- Parameters:
mode (int |
All) – The phonon mode for which to return the inelastic transmission. If not provided, the result for all modes will be returned. Default:All- Returns:
The symmetric negative inelastic transmission. If mode=All, the shape of the returned array is (number_of_degrees_of_freedom, number_of_energies, number_of_kpoints, number_of_qpoints, number_of_spins). If mode=integer, the shape is (number_of_energies, number_of_kpoints, number_of_qpoints, number_of_spins).
- Return type:
numpy.ndarray.
- inelasticTransmissionSymmetricPositiveBias(mode=None)¶
Return the symmetric inelastic transmission for each mode for positive bias voltages.
- Parameters:
mode (int |
All) – The phonon mode for which to return the inelastic transmission. If not provided, the result for all modes will be returned. Default:All- Returns:
The symmetric positive inelastic transmission. If mode=All, the shape of the returned array is (number_of_degrees_of_freedom, number_of_energies, number_of_kpoints, number_of_qpoints, number_of_spins). If mode=integer, the shape is (number_of_energies, number_of_kpoints, number_of_qpoints, number_of_spins).
- Return type:
numpy.ndarray.
- infinitesimal()¶
- Returns:
The infinitesimal used in self-energy calculations.
- Return type:
PhysicalQuantity of type energy.
- interactionRegion()¶
- Returns:
The atom indices for the interaction region.
- Return type:
list of ints
- kpoints()¶
- Returns:
The k-points.
- Return type:
list of floats with shape (K, 3), where K is the number of k-points.
- maximumPhononEnergy()¶
- Returns:
Return the maximally allowed phonon energy.
- Return type:
PhysicalQuantity of type energy.
- metatext()¶
- Returns:
The metatext of the object or None if no metatext is present.
- Return type:
str | None
- method()¶
- Returns:
The method used for the calculation.
- Return type:
XLOE|LOE
- minimumPhononEnergy()¶
- Returns:
The minimally allowed phonon energy.
- Return type:
PhysicalQuantity of type energy.
- nlinfo()¶
Information about the inelastic transmission spectrum analysis.
- Returns:
A dictionary containing the analysis information.
- Return type:
dict
- nlprint(stream=None)¶
Print a string containing an ASCII table useful for plotting the AnalysisSpin object.
- Parameters:
stream (python stream) – The stream the table should be written to. Default:
NLPrintLogger()
- phononEnergies()¶
- Returns:
The phonon energies for all the modes.
- Return type:
PhysicalQuantity of energy unit.
- phononModes()¶
- Returns:
The phonon mode indices.
- Return type:
list of ints
- qpoints()¶
- Returns:
The q-points.
- Return type:
list of floats with shape (Q, 3), where Q is the number of q-points.
- setMetatext(metatext)¶
Set a given metatext string on the object.
- Parameters:
metatext (str | None) – The metatext string to set. Use “None” to remove current metatext.
- uniqueString()¶
Return a unique string representing the state of the object.
Usage Examples¶
Calculate a InelasticTransmissionSpectrum. Notice that the DeviceConfiguration, the DynamicalMatrix and the HamiltonianDerivatives are read from a file filename.nc from a separate calculation.
# Example of inelastic transmission spectrum
# Load device configuration
device_configuration = nlread('filename.nc', DeviceConfiguration)[0]
# Load dynamical matrix
dynamical_matrix = nlread('filename.nc', DynamicalMatrix)[0]
# Load hamiltonian derivatives
hamiltonian_derivatives = nlread('filename.nc', HamiltonianDerivatives)[0]
# -------------------------------------------------------------
# Inelastic transmission spectrum
# -------------------------------------------------------------
inelastic_transmission_spectrum = InelasticTransmissionSpectrum(
configuration=device_configuration,
dynamical_matrix=dynamical_matrix,
hamiltonian_derivatives=hamiltonian_derivatives,
energies=numpy.linspace(0, 0, 1)*eV,
kpoints=MonkhorstPackGrid(1, 1),
qpoints=MonkhorstPackGrid(1, 1),
method=XLOE,
phonon_modes=All,
)
# -------------------------------------------------------------
# Post processing of data
# -------------------------------------------------------------
# Setup bias voltages
bias = numpy.linspace(-0.1, 0.1, 1000)*Volt
# Set temperature
temp = 10*Kelvin
# Calculate the inelastic current
I = inelastic_transmission_spectrum.inelasticCurrent(bias=bias, temperature=temp)
# Calculate the inelastic conductance
G = inelastic_transmission_spectrum.inelasticConductance(bias=bias, temperature=temp)
Large Device Calculations¶
In order to calculate the InelasticTransmissionSpectrum for a large device with thousands of atoms, it is possible to apply certain approximations that will speed up the calculations significantly.
Summing up phonon modes in energy intervals¶
The first option is to specify phonon_energy_intervals instead of phonon_modes=All like:
inelastic_transmission_spectrum = InelasticTransmissionSpectrum(
configuration=device_configuration,
dynamical_matrix=dynamical_matrix,
hamiltonian_derivatives=hamiltonian_derivatives,
phonon_energy_intervals=numpy.linspace(0, 0.1, 10)*eV,
)
For a device with, say, 1000 atoms in the central region, there will be 3000 phonon modes. However, by specifying phonon_energy_intervals the phonon modes will be summed in each interval to form new effective phonon modes. The number of intervals required will depend on the actual system, but typical values between 10 and 50 will often suffice, thus dramatically reducing the number of required calculations.
Note
When analyzing the results of an InelasticTransmissionSpectrum calculated with the phonon_energy_intervals parameter, the number of phonon modes one can query for will be the number of intervals used in the calculation.
In the above example, the intervals will be [0, 0.01]*eV, [0.01, 0.02]*eV, .... The i’th effective phonon mode corresponding to the energy interval \([\epsilon_i^0, \epsilon_i^1]\) will then be defined as (see also notes below)
Much fewer calculations then need to be performed in InelasticTransmissionSpectrum and the calculation time will be reduced very significantly for large devices.
Note
The use of phonon_energy_intervals is an approximation. It is intented to be used in large device calculations, where the primary focus is on the total current. It should not be used for, e.g., a molecular junction, where interest is on the inelastic signal from individual phonons.
Using bulk DynamicalMatrix and HamiltonianDerivatives¶
Many device calculations are performed for a structure which is translationally invariant in the C-direction, apart from doping profiles and electrostatic regions. This might be the case for a field effect transistor with a nin-juction or a pn-junction where the electrodes and the central region are composed of the same material.
In that case, it is possible to calculate the DynamicalMatrix and HamiltonianDerivatives for the smallest repeatable unit cell, provided that the central region atomic structure can be represented as:
central_region = bulk_configuration.repeat(nc=nc)
where nc is the number of times the small bulk configuration should be repeated in order to form the central region.
The calculation of the DynamicalMatrix and HamiltonianDerivatives for the small bulk_configuration will be much faster than the corresponding calculations for the full device configuration.
The InelasticTransmissionSpectrum can then be calculated as follows:
# Calculate the bulk hamiltonian derivatives for the small system.
bulk_hamiltonian_derivatives = HamiltonianDerivatives(bulk_configuration)
# Calculate the bulk dynamical matrix for the small system.
bulk_dynamical_matrix = DynamicalMatrix(bulk_configuration)
# Calculate the inelastic transmission spectrum using bulk dynamical matrix
# and bulk hamiltonian derivatives.
inelastic_transmission_spectrum = InelasticTransmissionSpectrum(
configuration=device_configuration,
bulk_dynamical_matrix=bulk_dynamical_matrix,
bulk_hamiltonian_derivatives=bulk_hamiltonian_derivatives,
phonon_energy_intervals=numpy.linspace(0, 0.1, 10)*eV,
)
By using both the phonon_energy_intervals, bulk_dynamical_matrix and bulk_hamiltonian_derivatives it is feasible to perform InelasticTransmissionSpectrum calculations for devices with several thousand of atoms.
Note
Using the bulk_dynamical_matrix and bulk_hamiltonian_derivatives relies on the assumption that the dynamical matrix and the derivative of the Hamiltonian are translational invariant, although the Hamiltonian itself is not. In, e.g., a pn-junction the Hamiltonian is not translational invariant due to the doping profile. However, it might be a reasonable approximation to assume that the derivative is approximately translational invariant thus justifying the use of bulk_hamiltonian_derivatives.
It is not possible to use bulk_dynamical_matrix and bulk_hamiltonian_derivatives for device configurations containing interfaces between different materials.
Notes¶
In order to calculate the InelasticTransmissionSpectrum three main ingredients are needed:
A DeviceConfiguration to get the Hamiltonian of the system.
A DynamicalMatrix object for calculating the phonon modes.
A HamiltonianDerivatives object for calculating the electron-phonon coupling matrix, \(M^\lambda\), for a particular phonon mode, \(\lambda\).
The device configuration used as input to InelasticTransmissionSpectrum must be exactly the same as used for the HamiltonianDerivatives and DynamicalMatrix objects. As explained above, for device configurations with a translationally invariant central region, it is possible to calculate the HamiltonianDerivatives and DynamicalMatrix objects for bulk configurations, which can be repeated to form the central region of the device.
From the dynamical matrix \(D\) we can obtain the phonon frequencies, \(\omega_\lambda\), and eigenvectors, \(\mathbf{u}^\lambda\),
The electron-phonon coupling matrix for a given phonon mode, \(\lambda\), is obtained as:
where
and where the sum runs over atom indices \(I\) and Cartesian directions \(\nu=(x,y,z)\). The derivative of the Hamiltonian is what is being calculated in the HamiltonianDerivatives object.
The theory behind the InelasticTransmissionSpectrum can be found in Refs. [1], [2]. In short, the starting point is the Meir-Wingreen current formula:
where \(G^{</>}\) are the lesser/greater Green’s functions for the central region (including electron-phonon self-energies) and \(\Sigma_{L}^{</>}\) are the lesser/greater self-energies due to coupling to the left electrode. The electron-phonon coupling self-energies are described in the self-consistent Born approximation (SCBA) assuming free (uncoupled to electrons and uncoupled to electrodes) phonon Green’s functions. The phonon spectral function is thus a series of delta-peaks at \(\pm \hbar\omega_\lambda\). We further assume that the phonon occupations are given by the Bose-Einstein distribution. Currently, it is thus not possible to include heating effects.
The SCBA electron-phonon self-energies are computationally demanding as they require an integral over energy, e.g. [2].
The idea behind the Lowest Order Expanssion (LOE) and eXtended LOE (XLOE) is to circumvent these integrals by assuming that the spectral function (or density of states) in the central region varies slowly on an energy scale given by the phonon energies, \(\hbar\omega\). By expanding the Meir-Wingreen current formula to the second order in M (this is the lowest order) one can derive the LOE current expression:
where \(I_0(V)\) is the non-interacting or elastic current that can be obtained from the normal transmission function. \(I_\lambda^{sym}\) and \(I_\lambda^{asym}\) are universal current functions given by
and
where \(\mathcal{H}_{\epsilon'}\) is the Hilbert transform and the bias is defined via \(eV = \mu_R - \mu_L\). The inelastic transmission functions \(\mathcal{T}_\lambda^{sym}\) and \(\mathcal{T}_\lambda^{asym}\) are given by
where \(H.c.\) denotes hermetian conjugate and where the spectral functions are
and
In the above equations, all the Greens function are non-interacting, i.e. they do not include the electron-phonon self-energies. When using LOE, all Green’s function, self-enegies, and spectral functions are evaluated at the same energy. In XLOE, certain Green’s functions and spectral functions will be evaluated at energies \(\epsilon_{\pm} = \epsilon \pm \hbar\omega_\lambda/2\). This means that many more calculations are needed for the XLOE calculations which therefore takes more time.
LOE or XLOE¶
The LOE is originally developed for studying molecular junctions where a molecule is placed between metallic electrodes. If there are no molecular states close to the Fermi energy, the LOE will be sufficient to use. However, if there are states close to the Fermi energy, the XLOE will be a more correct description as it allows some variation of the density of states. XLOE should also be used if transport occurs close to semi-conductor band edges.
In XLOE one distinguishes between phonon emission and phonon absorption processes, as illustrated in Fig. 180. The inelastic transmission functions for the emission process, corresponding to a positive bias can be obtained as:
t_sym_pos = inelastic_transmission_spectrum.inelasticTransmissionSymmetricPositiveBias()
t_asym_pos = inelastic_transmission_spectrum.inelasticTransmissionAsymmetricPositiveBias()
and for phonon absorption, corresponding to a negative bias situation as:
t_sym_neg = inelastic_transmission_spectrum.inelasticTransmissionSymmetricNegativeBias()
t_asym_neg = inelastic_transmission_spectrum.inelasticTransmissionAsymmetricNegativeBias()
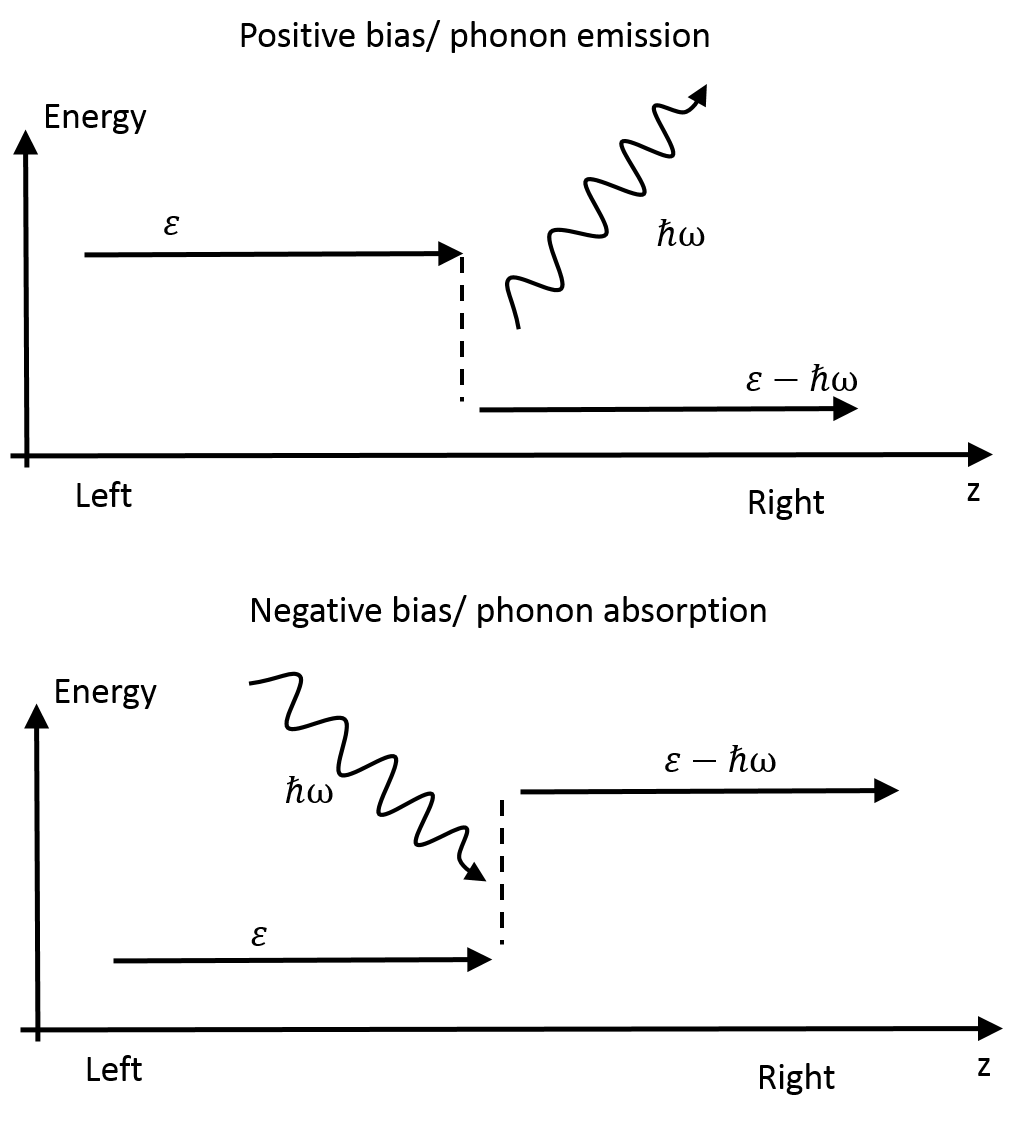
Fig. 180 In XLOE phonon emission and absorption processes are distinguished. The upper panel illustrates a phonon emission process, where an electron incident from the left electrode emits a phonon and exit the right electrode with a lower energy. At low temperatures, this process is only possible at positive bias voltages, hence the name. The lower panel illustrates a phonon absorption process where the electron exits with a higher energy in the right electrode than in the left.¶
How many energy points¶
The InelasticTransmissionSpectrum can be called with a user-defined list of energies (default is \(E=0\,eV\) , relative to the Fermi energy). If the non-interacting transmission is finite at the Fermi level (e.g. in a molecular junction with metallic leads) it will often be sufficint to calculate the inelastic transmisssion spectrum only at the Fermi energy and use the LOE current formula, which is called by:
bias = numpy.linspace(-0.1,0.1,100)*Volt
current = inelastic_transmission_spectrum.inelasticCurrent(bias=bias)
The conductance \(dI/dV\) and inelastic electron tunneling spectra (IETS) \(d^2I/dV^2\) can likewise be obtained from the LOE current expression as:
bias = numpy.linspace(-0.1,0.1,100)*Volt
conductance = inelastic_transmission_spectrum.inelasticConductance(bias=bias)
iets = inelastic_transmission_spectrum.inelasticElectronTunnelingSpectroscopy(bias=bias)
In situations where there are no electrode states at the Fermi energy, like in a low- or un-doped semi-conductor, the direct use of the LOE and XLOE expressions will result in zero inelastic current, when evaluated at the Fermi energy. In that case, the inelastic transmission spectra must be calculated at a range of energies (like the normal TransmissionSpectrum). The current can then be calculated as:
bias = numpy.linspace(-0.1,0.1,100)*Volt
current = inelastic_transmission_spectrum.inelasticCurrentIntegral(bias=bias)
In this case the current is calculated in a way, following Ref. [3]:
where the energy- and mode depended prefactors are
and
\(n_F\) and \(n_B\) are the Fermi-Dirac and Bose-Einstein distribution functions respectively.