SMI-Cluster Adsorption Energy Calculations¶
This application demonstrates automated and semi-automated workflows to calculate interaction energies between small-molecule inhibitors (SMIs) and metal clusters. SMIs are used for masking surface sites in area selective deposition (ASD), making understanding their interaction with metal surfaces critical for process optimization. The semi-automated workflow employs a sophisticated tag-based approach to identify anchor atoms and removable atoms, enabling automated complex generation and energy calculations. The fully automated workflow (applicable only for acid-derivative SMIs) executes all steps without manual tagging. Both workflows automatically identify unique ligands in clusters, prepare SMI molecules for adsorption, and form adsorption complexes to compute interaction energies.
Important
QuantumATK Version: This application is designed for QuantumATK X-2025.06.
Computational Method: This application uses ML potentials level of theory for geometry optimization and energy calculations to achieve accurate interaction energy predictions.
Workflow Options: Two execution modes available:
Semi-automated: Users manually tag groups to remove and anchor atoms
Fully automated: All processes executed automatically (applicable only for acid-derivative SMIs)
You can download the required files below:
AdsorptionEnergies_tagBasedScript.py- Semi-automated workflow scriptAdsorptionEnergies_TagBased_workflow.hdf5- Semi-automated workflowGenerateAdsorptionComplex.pye- Semi-automated compile modulesAcidDerivative_AdsorptionEnergies_script.py- Fully automated workflow scriptAcidDerivative_AdsorptionEnergies_workflow.hdf5- Fully automated workflowGenerateAcidDerivativeComplex.pye- Fully-automated compile modules
Note
All .pye files must be downloaded and placed in the same directory as the script or workflow file for proper execution.
Key Features
Two workflow modes: Semi-automated (manual tagging) or fully automated execution
Automatic ligand identification: Identifies all unique ligands in metal clusters
SMI preparation: Automatic removal of hydrogen from OH groups
Complex formation: Uses adsorption tool to create cluster-SMI complexes
Parallel execution: Array script support for efficient calculations
ML potential calculations: Accurate geometry optimization and energy calculations using machine learning potentials
Comprehensive output: Table with adsorption energies and all optimized geometries
Application Goal and Workflow¶
Objective¶
The goal of this application is to calculate interaction energies between metal clusters and small-molecule inhibitors (SMIs) with minimal human effort.
Input:
Metal cluster geometries
SMI molecule geometries
Output:
Table of adsorption energies for all cluster-SMI combinations
Optimized geometries of complexes, isolated clusters, and isolated SMIs
Automated Workflow Steps¶
The workflow executes the following steps:
Identify unique ligands in metal clusters (fully automated)
Remove ligand from cluster to create binding site (automated or manual tagging)
Prepare SMI molecule by removing hydrogen from OH group (automated or manual tagging)
Select anchor atom in SMI for adsorption (automated or manual tagging)
Form adsorption complex using adsorption tool with selected anchor atom
Optimize geometries of complex, cluster, and SMI using ML potentials
Calculate adsorption energies from energy differences
Generate output table with organized results
Scientific Background¶
Adsorption Energy for SMI-Cluster Interactions¶
The adsorption energy quantifies the strength of interaction between a small-molecule inhibitor (SMI) and a metal cluster:
Understanding SMI-cluster interactions is critical for ALD surface chemistry, particularly for area selective deposition (ASD), where SMIs are used to mask specific surface sites. Calculating these interaction energies enables prediction of surface modification, ligand exchange reactions, and the effectiveness of site-selective blocking for controlled film nucleation.
Computational Approach¶
The adsorption energy is calculated as:
where:
\(E_{\text{Complex}}\) is the total energy of the SMI-cluster adsorption complex
\(E_{\text{Cluster}}\) is the energy of the isolated cluster (with one ligand removed)
\(E_{\text{SMI}}\) is the energy of the isolated SMI molecule (with hydrogen removed from OH group)
Negative adsorption energy indicates favorable (exothermic) adsorption, while positive values indicate unfavorable (endothermic) interactions.
Automatic Identification of Unique Ligands¶
Critical Automation Step¶
The automatic identification of unique ligands in metal clusters is performed for all workflow modes. The script analyzes the cluster geometry and automatically determines which ligands are chemically unique, thereby eliminating redundant calculations.
Ligand Identification Process¶
The automated script performs unique ligand identification by:
Analyzing connectivity: Identifies all ligands bonded to the central metal atom(s)
Grouping by type: Groups chemically equivalent ligands together based on bond graph and graph isomorphism
Selecting representatives: Chooses one representative from each unique group for removal and complex formation
This ensures that only unique binding sites are considered, avoiding redundant calculations while capturing all distinct adsorption environments.
Workflow Modes¶
Semi-Automated Workflow¶
In the semi-automated mode, users provide manual tags to specify:
Which ligand to remove: Tag the specific ligand or ligand group for removal
Which hydrogen to remove from SMI: Tag the H atom in the OH group to be removed
Anchor atom selection: Tag the atom in SMI that will bind to the cluster
The workflow uses hardcoded tag names that must be applied in NanoLab:
# Required tag names for semi-automated workflow
"remove" # Tag for ligand/group to be removed from cluster
"remove" # Tag for hydrogen atom to be removed from SMI
"anchor" # Tag for atom in SMI that forms bond with cluster
Users must assign these exact tag names to the appropriate atoms in NanoLab before running the workflow.
Fully Automated Workflow¶
In the fully automated mode, the script handles all steps without manual tagging:
Automatic ligand selection: Identifies and removes one representative from each unique ligand type
Automatic hydrogen removal: Detects OH groups and removes hydrogen automatically
Automatic anchor selection: Identifies the optimal anchor atom (typically oxygen in OH group)
Complex Formation with Adsorption Tool¶
The workflow uses QuantumATK’s AdsorbedSurfaceGenerator (the backend functionality of the GUI adsorption tool) to create cluster-SMI complexes programmatically:
Process:
Position SMI near the binding site on the cluster
Orient SMI with anchor atom pointing toward the cluster
Set initial distance between anchor atom and binding site
Generate initial complex geometry
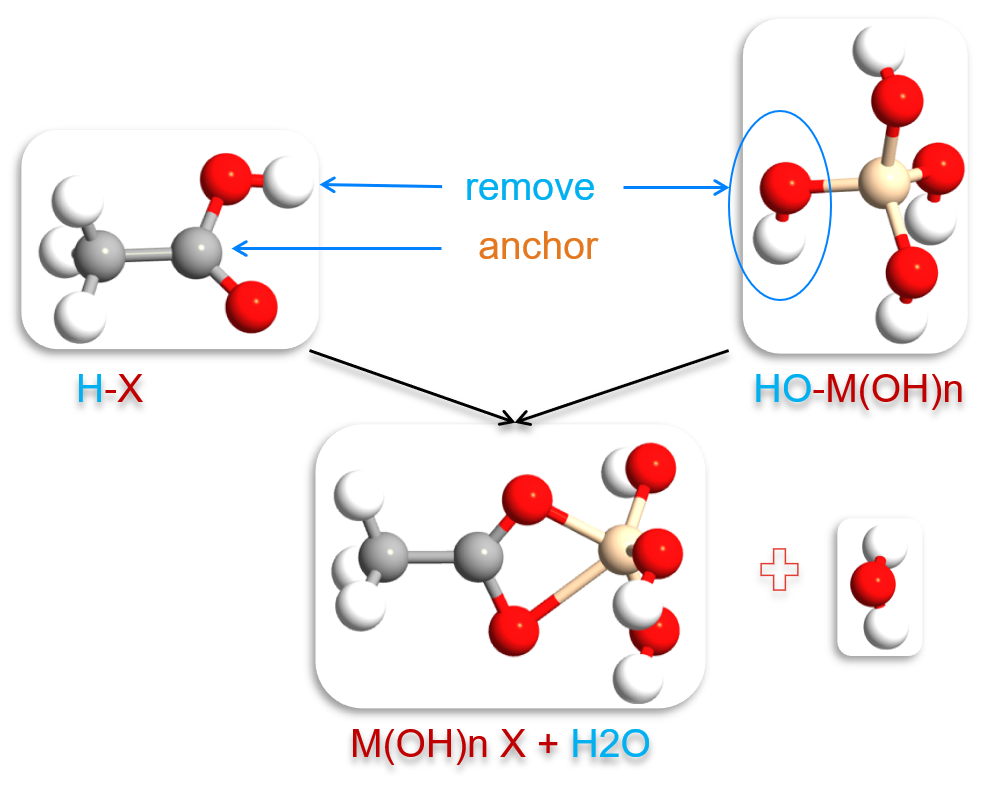
Fig. 154 Illustration of complex formation between a metal cluster (with ligand removed) and an SMI molecule. The anchor atom in the SMI is positioned at the binding site created by ligand removal.¶
The AdsorbedSurfaceGenerator ensures proper initial geometry for subsequent optimization, with the anchor atom positioned to form a bond with the cluster at the site where the ligand was removed.
Workflow¶
The workflow handles multiple metal clusters and SMIs systematically. The workflow script iterates over all combinations of metal clusters and SMIs, executing a systematic workflow with nested loops to calculate all combinations:
[2 Molecules: CH₃COOH, CH₃COSH] × [2 Clusters: Al(NH₂)₃, Si(OH)₄]
↓
[Generate 2×2 = 4 Combinations]
↓
For each combination:
├─ Align surface cluster
├─ Position molecule above surface
├─ Generate complex
└─ Create leaving fragment
↓
For each of 4 configurations (cluster, molecule, complex, fragment):
├─ Set calculator
├─ Optimize geometry
└─ Calculate total energy
↓
Compute E_ads = E(complex) + E(fragment) - E(cluster) - E(molecule)
↓
Store results with labels
↓
[Final Table: 4 rows with adsorption energies]
Output¶
The workflow generates a comprehensive table of adsorption energies:
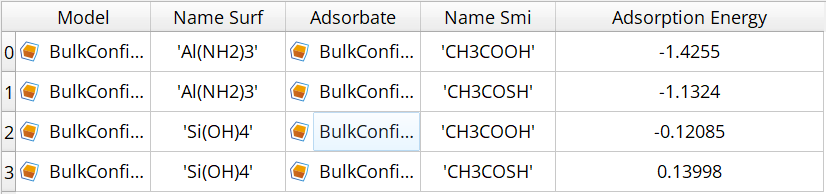
Fig. 155 Example output table showing adsorption energies for various cluster-SMI combinations with corresponding geometry files.¶
Summary¶
This application provides flexible workflows to calculate adsorption energies between metal clusters and small-molecule inhibitors (SMIs) relevant to area selective deposition. Starting from cluster and SMI geometries in NanoLab, the workflow automatically identifies unique ligands, prepares binding sites, and generates adsorption complexes using the AdsorbedSurfaceGenerator. Two modes are available: semi-automated (with manual atom tagging for precise control) and fully automated (for high-throughput screening of acid derivatives). All geometry optimizations and energy calculations employ accurate ML potentials, with support for parallel execution via array scripts. The output includes a comprehensive table of adsorption energies and optimized geometries for all cluster-SMI combinations, enabling efficient screening of SMI reactivity and surface blocking effectiveness.