Building a Graphene Nanoribbon Device¶
You will here learn how to build the device configuration used in the tutorial Transport in a graphene nanoribbon with a distortion.
The simplest way to build a device is to first build the desired scattering region as a bulk, including any defects and other deviations from perfect periodicity, and then use the Device from Bulk plugin to construct and attach the electrodes. Geometry optimization should also be considered; this can be done before and/or after the device is “assembled”.
Bulk Central Region¶
First step is to build a perfect (defect free) graphene nanoribbon. Open
the  Builder and use the Nanoribbon plugin to create a rather
narrow ribbon with chiral indices (2,2), corresponding to a zigzag nanoribbon,
8 carbon atoms wide. Then use the tool
to repeat the nanoribbon unit cell 12 times along the C direction.
Builder and use the Nanoribbon plugin to create a rather
narrow ribbon with chiral indices (2,2), corresponding to a zigzag nanoribbon,
8 carbon atoms wide. Then use the tool
to repeat the nanoribbon unit cell 12 times along the C direction.
Tip
Use the keyboard shortcut Ctrl+R to center the 3D view after applying the repeat operation.

Defect in the Scattering Region¶
Let us now create a Stone–Wales defect in the middle of the nanoribbon – in what will become the scattering region in the nanoribbon device.
Select the two atoms indicated in the figure below by holding down Ctrl while clicking them. Then use the tool to rotate them 90 degrees around the X axis. Keep the Rotate around selection center box checked while applying the rotation.

Device from Bulk¶
Next step is to build the actual device. Select the nanoribbon Stash item, and navigate to the plugin. The purpose of this plugin is to attach left and right electrodes to the defected central region created above.
The plugin automatically detects the periodicity of the region closest to the edges of the central region, and suggests valid electrodes. In order for this to work well, there should be at least one full period plus one additional layer of the electrode present in the central region.
Important
You should in general be careful not to make the central region too short along the C direction: It must be long enough to accomodate any defects you introduce as well as a perfectly periodic structure in both ends, where the central region meets the electrodes.
The electrodes should also not be too short: They must be able to include all relevant Hamiltonian matrix elements with the central region. A rough estimate is 7–8 Å in many systems.
Both points above relate to the concepts of electrode extensions and scattering region, which you can learn more about in the tutorials Transport calculations with QuantumATK and Transport in a graphene nanoribbon with a distortion.
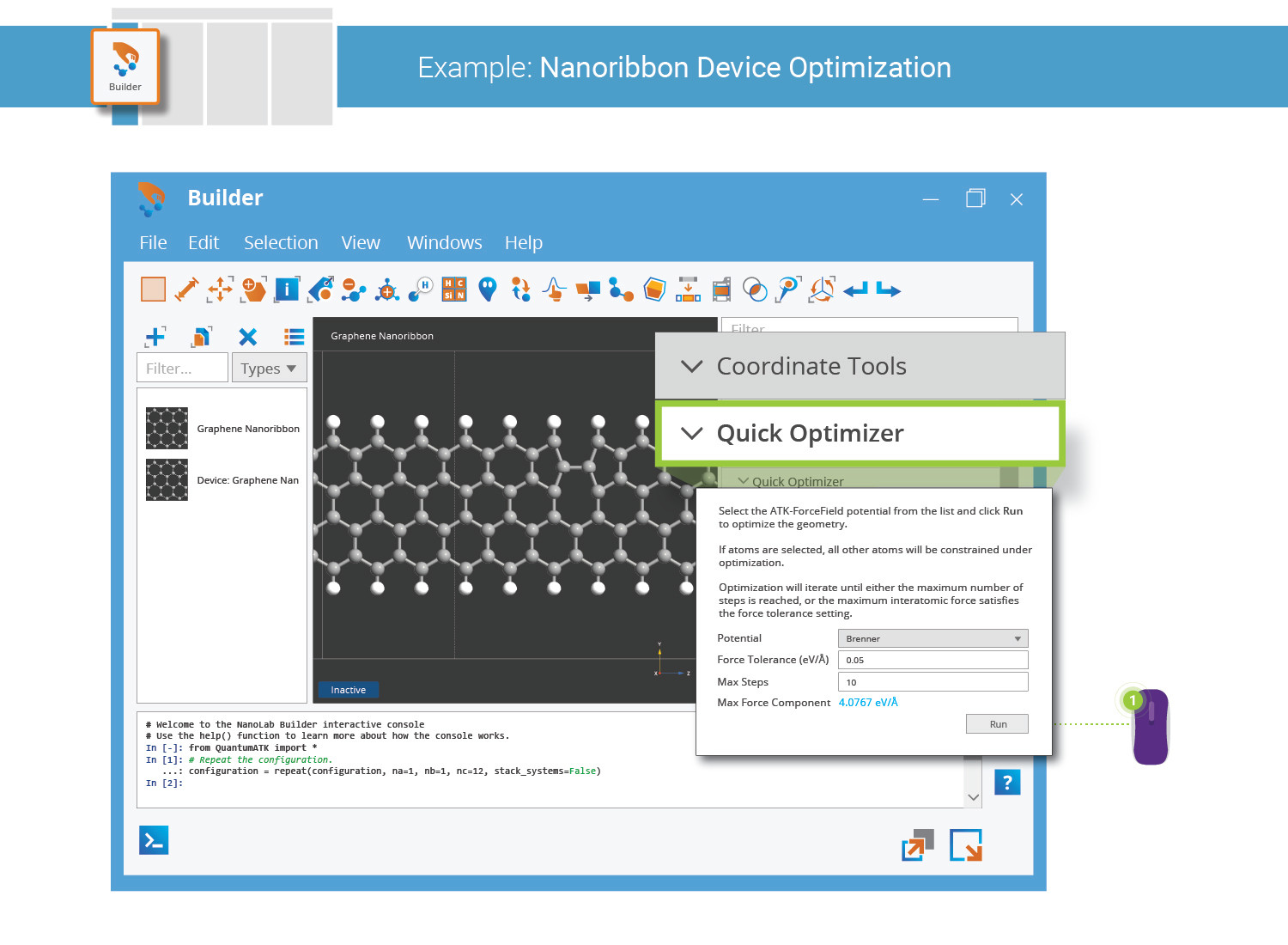
Optimizing the Geometry¶
The perfect nanoribbon central region was created without any geometry optimization. Although the C–C and C–H bond lengths are most likely very close to optimum far from the Stone–Wales defect, the local geometry around the defect certainly needs force minimization.
The Builder offers the very convenient Quick Optimizer plugin, which uses fast pair-potential calculations implemented in the ATK-ForceField calculator engine. Choose the Brenner potential and click Run to execute 10 geometry optimization steps.
Note
Although it may not be obvious to the naked eye, the electrode extensions in both ends of the central region are automatically constrained during a device geometry optimization. This retains their exact geometric correspondence to the neighboring electrode.
10 optimizer steps are not enough to fully minimize the forces. Increase the maximum number of steps to 100 and run one more geometry optimization to reach full convergence.
Tip
The Quick Optimizer can also be used to pre-minimize the forces in a configuration prior to performing a relaxation with DFT, which is computationally more heavy. However, beware that high-quality classical potentials are not available for all possible combinations of elements.