RangeSeparatedDensityPredictor¶
Included in QATK.Calculators.DFT, QATK.MLDFT
- class RangeSeparatedDensityPredictor(model=None, probe_count=None, gpu_acceleration=None)¶
Class for parameters needed for a density predictor using range-separated density models.
- Parameters:
model (GridValuesModel) – Class specifying the path to the density model.
probe_count (int, optional) – Number of probe graph nodes to use, Default:
5000.gpu_acceleration (Enabled | Disabled | Automatic, optional) – Whether to use GPU acceleration. If set to
Automatic, the predictor will use the GPU if available. Default:Automatic
- densityModel()¶
Get the density model used by this predictor. Deprecated, use model() instead.
- Returns:
The density model.
- Return type:
- gaussianSigma()¶
Get the Gaussian sigma used for separating the short-range and long-range parts of the density.
Gaussian sigma is always taken from the long-range model, even though we use it to convolute the short-range part. It is physically meaningless to have two different sigmas (and if the full model is used instead of a short-range model, there is no sigma at all), so we choose to ignore sigma from the short-range model.
- Returns:
The Gaussian sigma in Bohr.
- Return type:
float
- getDeepDFTModels()¶
Get the DeepDFT models for the short-range and long-range parts.
- getValidatedModelDirs()¶
Validate that the model directory contains exactly two subdirectories for the short-range and long-range models.
- Raises:
NLValueError – If the model directory does not contain exactly two subdirectories.
- Returns:
List of absolute paths to the model directories.
- Return type:
list[str]
- gpuAcceleration()¶
Return whether to use CUDA-supported GPU acceleration.
- Returns:
The GPU acceleration flag.
- Return type:
Enabled | Disabled | Automatic
- longRangeElectronDifferenceDensity(configuration, calculator, ground=None)¶
Use the defined model to predict the long-range part of the electron difference density of a configuration :param configuration: The configuration for which to predict the electron density. :type configuration: MoleculeConfiguration | BulkConfiguration | DeviceConfiguration
- Parameters:
calculator (Calculator) – The calculator used to predict the electron density.
ground (bool) – Boolean controlling if the average electron difference density should be forced to zero.
- Returns:
The predicted long-range part of the electron density.
- Return type:
NLEngine.MPISharedScalarField
- model()¶
Return the parameters for the ml model.
- Returns:
The model parameters.
- Return type:
- nlinfo()¶
- Returns:
The nlinfo.
- Return type:
dict
- predictElectronDensity(configuration, calculator, ground=None)¶
Use the defined model to predict the electron density of a configuration.
- Parameters:
configuration (MoleculeConfiguration | BulkConfiguration | DeviceConfiguration) – The configuration for which to predict the electron density.
calculator (Calculator) – The calculator used to predict the electron density.
ground (bool) – Boolean controlling if the average electron difference density should be forced to zero.
- Returns:
The predicted electron density.
- Return type:
NLEngine.MPISharedScalarField
- predictElectronDifferenceDensity(configuration, calculator, ground=None)¶
Use the defined model to predict the electron difference density of a configuration.
- Parameters:
configuration (MoleculeConfiguration | BulkConfiguration | DeviceConfiguration) – The configuration for which to predict the electron density.
calculator (Calculator) – The calculator used to predict the electron density.
ground (bool) – Boolean controlling if the average electron difference density should be forced to zero.
- Returns:
The predicted electron density.
- Return type:
NLEngine.MPISharedScalarField
- predictElectronDifferenceDensityFromModel(deep_dft_model, configuration, calculator, ground=None)¶
Use the given model to predict the electron difference density of a configuration.
- Parameters:
deep_dft_model (DeepDFTModel) – The deep DFT model to use for the prediction.
configuration (MoleculeConfiguration | BulkConfiguration | DeviceConfiguration) – The configuration for which to predict the electron density.
calculator (Calculator) – The calculator used to predict the electron density.
ground (bool) – Boolean controlling if the average electron difference density should be forced to zero. Default:
True
- Returns:
The predicted electron density.
- Return type:
NLEngine.MPISharedScalarField
- probeCount()¶
Return the number of probes to use for the grid prediction.
- Returns:
The number of probes.
- Return type:
int
- shortRangeElectronDifferenceDensity(configuration, calculator, ground=None)¶
Use the defined model to predict the short-range part of the electron difference density of a configuration.
- Parameters:
configuration (MoleculeConfiguration | BulkConfiguration | DeviceConfiguration) – The configuration for which to predict the electron density.
calculator (Calculator) – The calculator used to predict the electron density.
ground (bool) – Boolean controlling if the average electron difference density should be forced to zero.
- Returns:
The predicted short-range part of the electron density.
- Return type:
NLEngine.MPISharedScalarField
- uniqueString()¶
Return a unique string representing the state of the object.
Notes¶
Similar to DensityPredictor, the machine learned RangeSeparatedDensityPredictor can significantly speed up the convergence of the self-consistent field (SCF) procedure in density functional theory (DFT) calculations for highly polarizable cases, surfaces, or ionic systems, where long-range electrostatic effects and nonlocal physics are important. The predictor provides an initial guess for the electron density, which can reduce the number of iterations required to reach convergence.
The way to use a range-separated density predictor in a DFT calculations is nearly identical to the standard DensityPredictor. One creates an instance of the RangeSeparatedDensityPredictor class and passes it to the AlgorithmParameters used in the DFT calculation. For example,
model = PretrainedGridValuesModels.density_HKMG_PBEHalf_PD_Medium_Y2026
grid_values_predictor = RangeSeparatedDensityPredictor(
model=model,
)
algorithm_parameters = AlgorithmParameters(grid_values_predictor=grid_values_predictor)
where PretrainedGridValuesModels.density_HKMG_PBEHalf_PD_Medium_Y2026 is a pre-trained density model included in QuantumATK.
Application of a range-separated density predictor is supported for both CPU-and GPU-based DFT calculations in QuantumATK. However, note that running on a GPU is significantly faster.
Use of a range-separated density predictor is supported for both LCAO and Plane Wave DFT calculations in QuantumATK, but currently only for normconserving pseudopotentials.
MGGA functionals cannot be used together with the density predictor. Other XC funcitonals are supported, but we recommend using the same XC functional as used during training of the density model for best results.
Pretrained models¶
QuantumATK includes a single pre-trained range-separated density predictor that can be used directly in DFT calculations:
- density_HKMG_PBEHalf_PD_Medium_Y2026 (Y-2026)
Model name: PretrainedGridValuesModels.density_HKMG_PBEHalf_PD_Medium_Y2026
Description: Specific model developed for simulations of HKMG stacks with structures like Si-SiO2-HfO2-TiN. Both SiO2, HfO2, and part of TiN layers can be amorphous. The model is trained with individual layers and explicit stacks containing Si, SiO2, HfO2, and TiN as well as Al dopants in HfO2. The model can be used to accelerate SCF convergence in DFT calculations of these gate stacks and similar structures and might even be used in non-selfconsistent calculations of e.g. the Projected Density of States (PDOS).
Pseudopotential: PseudoDojo
Basis set: Medium
Exchange correlation: PBE-1/2
Spin: Unpolarized
Usage Examples¶
Use the pre-trained density_HKMG_PBEHalf_PD_Medium_Y2026 model for the initial density in a DFT calculation. For this example
range_separated_density_predictor_example.py the number of SCF steps is
reduced from 37 to 25 by using the pre-trained density_HKMG_PBEHalf_PD_Medium_Y2026 model as compared to the default neutral atom initial density.
The training set for this model included the full HkMG stacks, in this example range_separated_density_predictor_HkMG_stack.py,
the SCF failed to converge within 100 steps when using the default neutral atom initial density,
while using the pre-trained density_HKMG_PBEHalf_PD_Medium_Y2026 model led to convergence in 30 SCF steps.
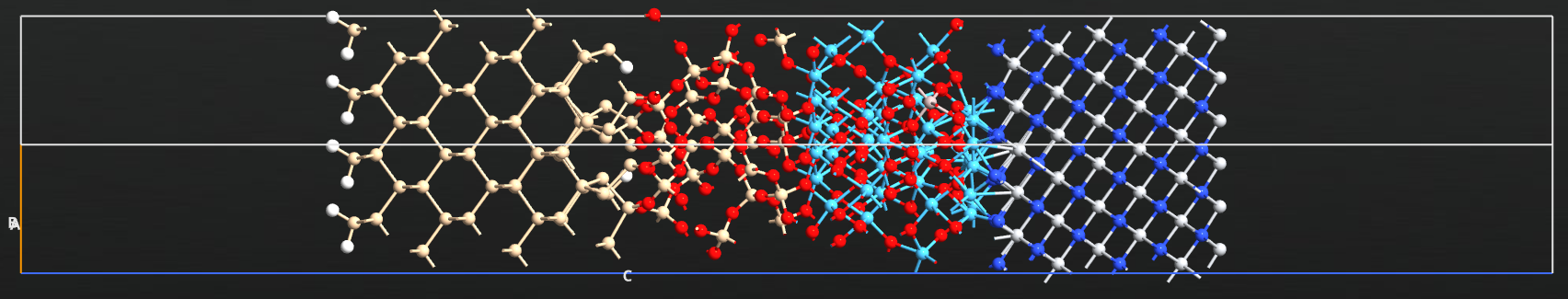
In the next example range_separated_density_predictor_HkMG_stack_nonscf.py,
we substituted an atom in the HfO layer in the HKMH stack with an aluminium atom to simulate doping of the HfO2 layer.
The structure is shown in the figure above.
We obtain very good results for the HartreeDifferencePotential and ProjectedDensityOfStates even without performing
self-consistent calculations, by using the predicted density directly in a non-self-consistent DFT calculation.
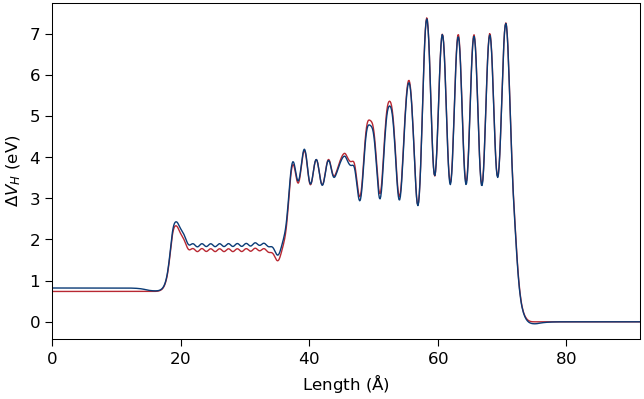
Illlustration of the HartreeDifferencePotential obtained from a non-self-consistent DFT calculation using the predicted density (red curve) and from the self-consistent calculation (blue curve) for a HKMG stack with an Al substitution in the HfO2 layer.
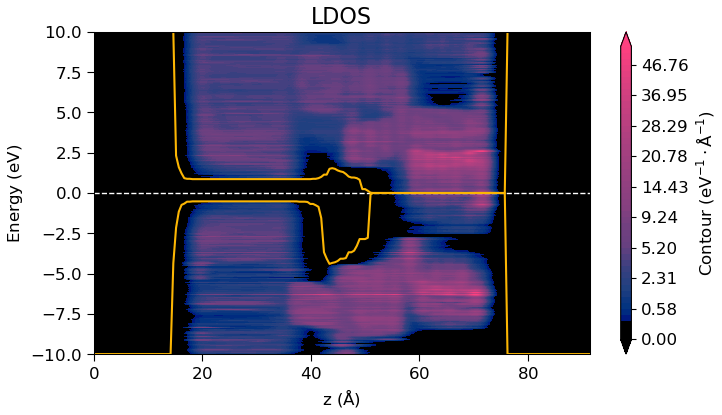
Illlustration of the ProjectedDensityOfStates obtained from a non-self-consistent DFT calculation using the predicted density.